
Vol. 24 - Num. 96
Colaboraciones especiales
El síndrome de Beckwith Wiedemann en la consulta de Atención Primaria. Ejemplo de la importancia del asociacionismo en las enfermedades raras
Elena Bermúdez de Castro Lópeza
aPediatra. CS San Blas. Parla. Madrid. España.
Correspondencia: E Bermúdez. Correo electrónico: elena.bermudez@salud.madrid.org
Cómo citar este artículo: Bermúdez de Castro López E. El síndrome de Beckwith Wiedemann en la consulta de Atención Primaria. Ejemplo de la importancia del asociacionismo en las enfermedades raras . Rev Pediatr Aten Primaria. 2022;24:429-34.
Publicado en Internet: 29-12-2022 - Número de visitas: 13327
Resumen
El síndrome de Beckwith Wiedemann es una enfermedad poco frecuente que tiene una prevalencia de 1 por cada 10 340 personas. Se caracteriza por presentar macrosomía, macroglosia, defectos en la pared abdominal, organomegalia, hipercrecimiento lateralizado y un riesgo aumentado de desarrollo de tumores en los primeros años de vida. En este artículo se presenta un protocolo actualizado para el manejo de esta patología en la consulta de Atención Primaria. Por otra parte, este síndrome nos sirve como ejemplo para ilustrar el importante papel que cumplen las asociaciones de pacientes en prestar apoyo a estas personas y a sus familiares, así como en incrementar la visibilidad de las enfermedades raras e incentivar la investigación.
Palabras clave
● Enfermedades raras ● Macroglosia ● Síndrome de Beckwith WiedemannLas enfermedades raras o poco frecuentes son aquellas que se presentan en menos de 5 por cada 10 000 habitantes. Esta cifra, que aisladamente parece muy baja, nos puede hacer pensar que muy pocas personas las padecen. Pero si consideramos que se han descrito unas 7000 patologías que cumplen esta característica, nos daremos cuenta de que un porcentaje importante de la población estaría implicado. Se calcula que en España unos 3 millones de personas tienen alguna de ellas; y, extrapolando a nivel mundial, un 6-8% de la población podría estar afectada. El 80% son de origen genético y la mayoría de ellas se presentan desde la infancia, pero otras no se detectan hasta la edad adulta. Muchas de ellas son crónicas y tienen una alta morbilidad y mortalidad1-3.
Actualmente disponemos de multitud de recursos para buscar información sobre las enfermedades raras. Además, la sensibilización de la sociedad y de las autoridades sanitarias con respecto a estas patologías es cada vez mayor, siendo las políticas sociales referentes a este tema cada vez más frecuentes1. No obstante, todavía queda mucho camino por recorrer. Debido a la baja incidencia, la investigación no resulta rentable y esto hace que los conocimientos y las posibilidades diagnósticas y terapéuticas que tenemos sobre muchas de estas enfermedades sean escasos. Por ello, no es infrecuente que el diagnóstico se retrase.
Las personas que las presentan se encuentran a diario con muchas dificultades, no solo por las consecuencias físicas que implican, sino también por los problemas psicológicos. Ansiedad, miedo, rechazo, aislamiento, soledad e incomprensión son sentimientos muy comunes entre estas personas. Se deduce, por tanto, que la calidad de vida de los pacientes y sus familias se resiente de manera importante, con multitud de problemas escolares y laborales.
En muchas ocasiones estos sentimientos mejoran cuando los afectados comparten sus experiencias con otras personas que han pasado por la misma situación. Una manera de localizar a estas personas es recurrir a las asociaciones de pacientes. Actualmente, en España hay más de 300 asociaciones de pacientes con enfermedades raras registradas que cumplen una función muy importante, ya que proporcionan información, orientación y apoyo psicológico a los pacientes recién diagnosticados. Además, tras recopilar las reivindicaciones de pacientes y sus familiares, pueden hacer de intermediarios entre los pacientes, la sociedad y diferentes instituciones gubernamentales a diferentes niveles para reivindicar derechos sanitarios y de cuidados. De esta forma se logra dar impulso a diferentes actividades, planes y estrategias basadas en las necesidades de los propios pacientes1-3. En 2009, la Federación Española de Enfermedades Raras (FEDER) realizó un estudio (ENSERio) que actualizó en 2017, con el que se comprobó que esta labor de las asociaciones repercute de manera positiva en el nivel de bienestar a varios niveles: económico, laboral, relacional, educativo, salud, ocio y tiempo libre3.
Los pediatras de Atención Primaria (PAP) debemos estar familiarizados con el manejo de estas patologías. En bastantes ocasiones somos los primeros que sospechamos la posible existencia de una enfermedad poco frecuente y, desde nuestra posición en primera línea, tenemos la capacidad de hacer un seguimiento del paciente a lo largo de todo su desarrollo desde un punto de vista biopsicosocial y en coordinación con otros niveles asistenciales.
Para ilustrar todo lo referido anteriormente, me gustaría poner como ejemplo el síndrome de Beckwith Wiedeman, que se considera una enfermedad poco frecuente porque su prevalencia es de 1 por cada 10 340 personas4. Se produce por diferentes alteraciones genéticas y epigenéticas complejas en el cromosoma 11p15.5. En esta región se encuentran varios genes que codifican proteínas implicadas en el crecimiento celular. Algunas de ellas, como la CDKN1C, frenan el crecimiento, y otras, por el contrario, lo estimulan, como el IGF2. En situaciones normales existe un equilibrio en la función de estas proteínas y el resultado es, por tanto, un crecimiento normal. En el caso del síndrome de Beckwith Wiedemann, hay un disbalance y predomina el papel de las proteínas potenciadoras del crecimiento, lo que conlleva la aparición de los síntomas característicos de esta patología: macrosomía, magroglosia, defectos en la pared abdominal, organomegalia, hipoglucemia neonatal secundara a hiperinsulinismo, hipercrecimiento lateralizado y aumento del riesgo de desarrollo de tumores embrionarios durante los primeros años de vida, principalmente el tumor de Wilms y el hepatoblastoma. También pueden presentar nevus flammeus, pliegues o hendiduras en los pabellones auriculares, y, con menos frecuencia, alteraciones cardiacas y renales. El desarrollo cognitivo es normal en la mayoría de los casos, aunque la prematuridad, la hipoglucemia neonatal severa y persistente o determinadas alteraciones genéticas pueden producir clínica neurológica4. La sintomatología es variable en función de la genética y del nivel de mosaicismo (del porcentaje de células afectadas), por lo que actualmente es más correcto hablar de espectro de Beckwith Wiedemann (BWsp)4-9 (Tabla 1).
| Tabla 1. Clínica y diagnóstico del espectro de Beckwith-Wiedemann4 |
|---|
| Clínica cardinal |
|
2 puntos por cada ítem:
|
| Clínica sugestiva |
|
1 punto por cada ítem:
|
| Diagnóstico clínico |
|

Debido al carácter multisistémico del BWsp, el manejo debe ser multidisciplinar y correctamente coordinado. El PAP, junto con diferentes médicos especialistas, realizará un adecuado seguimiento de estas personas durante todo su desarrollo para valorar las posibles complicaciones e indicar diferentes tratamientos en caso de ser necesario4,5. Ya desde el periodo prenatal, los obstetras y neonatólogos deben estar prevenidos si en las ecografías del embarazo se detectan alteraciones como macroglosia, macrosomía, onfalocele o placentomegalia, entre otras. Si esto ocurre, es aconsejable que el parto se realice en un centro con experiencia en esta patología y que, tras el nacimiento, manejen correctamente las posibles complicaciones, como la hipoglucemia o la cirugía del onfalocele si se diera el caso. Posteriormente, hay que vigilar la macroglosia, ya que esta puede dificultar la respiración, la alimentación, el lenguaje y ocasionar problemas ortodóncicos. El maxilofacial valorará la necesidad de cirugía para reducir el tamaño de la lengua y el ortodoncista, llegado el momento, podrá recomendar el uso de ortodoncia para corregir la malposición dentaria. La dismetría de extremidades inferiores secundaria al hipercrecimiento lateralizado puede dificultar la marcha o provocar escoliosis. El traumatólogo y el ortopeda recomendarán diferentes tratamientos según el grado de afectación.
Por otra parte, el posible desarrollo de tumores durante la infancia y niñez es la característica que más limita la calidad de vida de estas personas. En general, el riesgo tumoral en estos niños es del 8%4. El más común es el tumor de Wilms (52%), seguido del hepatoblastoma (14%) y del neuroblastoma (10%). En los últimos años se ha visto que el riesgo varía dependiendo de la alteración genética que presenten:
- Si la alteración es una hipermetilación en IC1 (H19/IGF2:IG-DMR) (que ocurre en un 5% de los pacientes), el riesgo tumoral es del 28%.
- Cuando presentan una hipometilación en IC2 (KCNQ1OT1:TSS-DMR) (50% de los pacientes con BWsp), el riesgo es del 2,6%.
- En los casos de disomía uniparental paterna (frecuencia de 20%), el riesgo es del 16%.
- Si tienen una alteración en CDKN1C (5%), el riesgo de aparición de tumores es del 6,9%.
Debido a esto es importante realizar un screening tumoral durante los primeros años de vida, que es el momento de mayor riesgo, adaptándolo a la alteración genética detectada. Según el consenso internacional publicado en 2018, se debe realizar una ecografía abdominal trimestral en todos los casos, excepto en los que presenten una hipometilación en IC2 aislada, ya que es la variante con un riesgo menor. En estos pacientes solo se hará el cribado si vemos algún dato de alarma en la anamnesis o en la exploración. No obstante, este es un tema controvertido que continúa revisándose, por lo que la decisión de realizar el screening en los pacientes con esta alteración concreta debe ser individualizada teniendo en cuenta la clínica, el criterio médico y la opinión de la familia4,5,9-12.
No debemos olvidar la posible afectación psicológica. El hecho de convivir con una enfermedad crónica puede tener un impacto importante a nivel afectivo y en el desarrollo psicosocial. Por tanto, deberemos siempre indagar en la necesidad de buscar apoyo de un profesional en salud mental, tanto para el niño como para la familia13.
Habitualmente, las características del BWsp son menos evidentes en las personas adultas y los rasgos faciales típicos que veíamos en los niños prácticamente no se aprecian. De todas formas, el médico de familia debe estar pendiente de algunos aspectos en estos adultos, como, por ejemplo, de las posibles secuelas de las cirugías que hayan precisado de pequeños. Asimismo, se han descrito problemas renales en adultos con BWsp, problemas auditivos e infertilidad. La incidencia de tumores es baja, pero, debido a la falta de estudios concluyentes, debemos estar alerta por si aparece algún dato de alarma sugestivo de la presencia de un tumor. Y, por último, debemos ofrecer consejo genético, principalmente en los casos con herencia familiar14.
Existen varias asociaciones de familias del BWsp a nivel europeo, gracias a las cuales en los últimos años ha aumentado la visibilidad de este síndrome y se ha fomentado la investigación. El pasado mes de junio se celebró en Cervia (Italia), el primer congreso internacional sobre el síndrome de Beckwith Wiedemann dirigido a familias y profesionales sanitarios. En él se reunieron médicos de varios países europeos (Italia, España, Reino Unido, Polonia), de Estados Unidos y Canadá. Se pudo llevar a cabo gracias al esfuerzo y al trabajo en equipo de varias asociaciones de pacientes (de Italia, España, Rumanía, Hungría y Suecia). Los diferentes especialistas pusieron en común los resultados de las últimas investigaciones sobre el BWsp y fue una buena oportunidad para establecer lazos que permitan continuar con la investigación a mayor escala. Por otra parte, las familias pudieron informarse, compartir su experiencia y resolver dudas, así como trasladar sus inquietudes a los profesionales sanitarios.
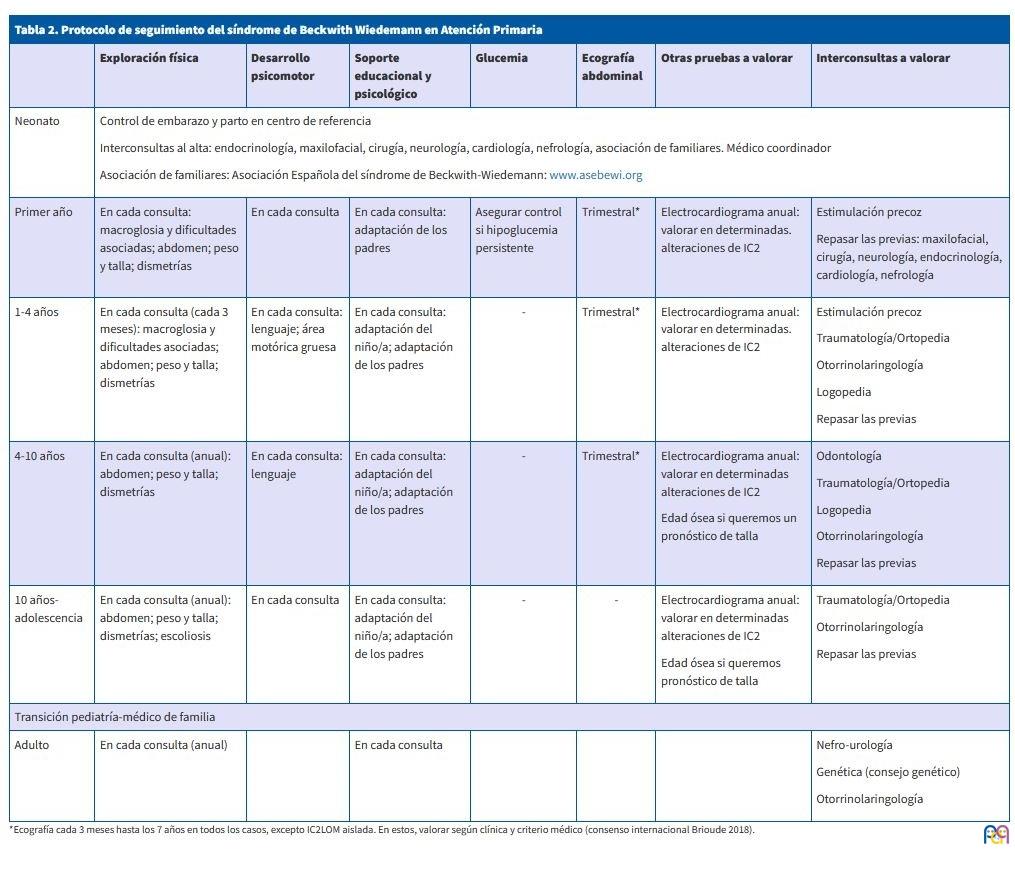
En 2014 se publicó en esta misma revista un protocolo de seguimiento de los pacientes con síndrome de Beckwith Wiedemann en Atención Primaria15. En él se resumen las exploraciones, pruebas diagnósticas y periodicidad con que deben realizarse, así como los especialistas a los que quizá sea necesario consultar. También se menciona la asociación de familias con la que pueden contactar. Desde su publicación, los avances en el estudio de esta enfermedad han sido llamativos y, por este motivo, he actualizado el protocolo (Tabla 2). Como principal novedad destaca la adaptación de las pruebas diagnósticas al tipo de alteración genética (según el consenso internacional publicado en 2018)4 y la mención de la transición desde el pediatra al médico de familia y del seguimiento por parte de este profesional. Este protocolo nos puede servir de guía en nuestra consulta de Atención Primaria (AP) pero siempre lo debemos adaptar a cada paciente particular.
| Tabla 2. Protocolo de seguimiento del síndrome de Beckwith Wiedemann en Atención Primaria | |||||||
|---|---|---|---|---|---|---|---|
| Exploración física | Desarrollo psicomotor | Soporte educacional y psicológico | Glucemia | Ecografía abdominal | Otras pruebas a valorar | Interconsultas a valorar | |
| Neonato |
Control de embarazo y parto en centro de referencia Interconsultas al alta: endocrinología, maxilofacial, cirugía, neurología, cardiología, nefrología, asociación de familiares. Médico coordinador Asociación de familiares: Asociación Española del síndrome de Beckwith-Wiedemann: www.asebewi.org |
||||||
| Primer año | En cada consulta: macroglosia y dificultades asociadas; abdomen; peso y talla; dismetrías | En cada consulta | En cada consulta: adaptación de los padres | Asegurar control si hipoglucemia persistente | Trimestral* | Electrocardiograma anual: valorar en determinadas. alteraciones de IC2 |
Estimulación precoz Repasar las previas: maxilofacial, cirugía, neurología, endocrinología, cardiología, nefrología |
| 1-4 años | En cada consulta (cada 3 meses): macroglosia y dificultades asociadas; abdomen; peso y talla; dismetrías | En cada consulta: lenguaje; área motórica gruesa | En cada consulta: adaptación del niño/a; adaptación de los padres | - | Trimestral* | Electrocardiograma anual: valorar en determinadas. alteraciones de IC2 |
Estimulación precoz Traumatología/Ortopedia Otorrinolaringología Logopedia Repasar las previas |
| 4-10 años | En cada consulta (anual): abdomen; peso y talla; dismetrías | En cada consulta: lenguaje | En cada consulta: adaptación del niño/a; adaptación de los padres | - | Trimestral* |
Electrocardiograma anual: valorar en determinadas alteraciones de IC2 Edad ósea si queremos un pronóstico de talla |
Odontología Traumatología/Ortopedia Logopedia Otorrinolaringología Repasar las previas |
| 10 años-adolescencia | En cada consulta (anual): abdomen; peso y talla; dismetrías; escoliosis | En cada consulta | En cada consulta: adaptación del niño/a; adaptación de los padres | - | - |
Electrocardiograma anual: valorar en determinadas alteraciones de IC2 Edad ósea si queremos pronóstico de talla |
Traumatología/Ortopedia Otorrinolaringología Repasar las previas |
| Transición pediatría-médico de familia | |||||||
| Adulto | En cada consulta (anual) | En cada consulta |
Nefro-urología Genética (consejo genético) Otorrinolaringología |
||||
Para concluir, quisiera recalcar la necesidad de que el PAP esté sensibilizado y bien formado en el manejo de las enfermedades poco frecuentes y que conozca el importante papel de las asociaciones de familias para así poder informar adecuadamente a sus pacientes. Sería conveniente incentivar la creación de protocolos similares al que aquí se presenta para mejorar la calidad de la atención a nuestros pacientes con estas patologías.
CONFLICTO DE INTERESES
Los autores declaran la ausencia de conflicto de intereses en la redacción del presente artículo.
ABREVIATURAS
BWsp: espectro de Beckwith Wiedemann. CDKN1C: cyclin dependent kinase inhibitor 1C. IC1: imprinting center 1 del cromosoma 11p15. IC2: imprinting center 2 del cromosoma 11p15. IGF2: factor de crecimiento insulínico tipo 2 (Insulin-like growth factor 2). PAP: pediatras de Atención Primaria.
BIBLIOGRAFÍA
- Estrategia en Enfermedades Raras del Sistema Nacional de Salud. Ministerio de Sanidad, Servicios Sociales e Igualdad. Actualización aprobada por el Consejo Interterritorial del Sistema Nacional de Salud el 11 de junio de 2014 [en línea] [consultado el 21/12/2022]. Disponible en www.sanidad.gob.es/organizacion/sns/planCalidadSNS/pdf/Estrategia_Enfermedades_Raras_SNS_2014.pdf
- Estado de situación del movimiento asociativo en enfermedades raras en España [en línea] [consultado el 21/12/2022]. Disponible en www.enfermedades-raras.org/que-hacemos/por-la-investigacion/obser/estudios/informes-especificos/estado-situacion-movimiento-asociativo-er-espna
- Estudio sobre situación de Necesidades Sociosanitarias de las personas con Enfermedades Raras en España, Estudio ENSERio. Federación Española de Enfermedades Raras (FEDER). Octubre de 2009 [en línea] [consultado el 21/12/2022]. Disponible en https://sid-inico.usal.es/documentacion/estudio-sobre-situacion-de-necesidades-sociosanitarias-de-las-personas-con-enfermedades-raras-en-espana-estudio-enserio/
- Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: An international consensus statement. Nat Rev Endocrinol. 2018;14:229-49.
- Duffy KA, Cielo CM, Cohen JL, Gonzalez Gandolfi CX, Griff JR, Hathaway ER, et al. Characterization of the Beckwith-Wiedemann spectrum: Diagnosis and management. Am J Med Genet C Semin Med Genet. 2019;181:693-708.
- Kim HY, Shin CH, Lee YA, Shin CH, Kim GH, Ko JM. Deciphering epigenetic backgrounds in a Korean Cohort with Beckwith- Wiedemann Syndrome. Ann Lab Med. 2022;42:668-77.
- Cammarata Scalisi F, Avendaño A, Stock F, Callea M, Sparago A, Riccio A, et al. Beckwith-Wiedemann syndrome. Clinical and etiopathogenic aspects of a model genomic imprinting entity. Arch Argent Pediatr. 2018;116:368-73.
- Zhang M, Sun C, Liu R, Dong C, Cheng R, Zheng Z, et al. Phenotypes and epigenetic errors in patients with Beckwith-Wiedemann syndrome in China. Transl Pediatr. 2020;9:653-61.
- Wang KH, Kupa J, Duffy KA, Kalish JM. Diagnosis and Management of Beckwith-Wiedemann Syndrome. Front Pediatr. 2020;7:562.
- Mussa A, Duffy KA, Carli D, Ferrero GB, Kalish JM. Defining an optimal time window to screen for hepatoblastoma in children with Beckwith- Wiedemann syndrome. Pediatr Blood Cancer. 2018;66:e27492.
- Mussa A, Duffy KA, Carli D, Griff JE, Fagiano R, Kupa J, et al. The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J Cancer Res Clin Oncol. 2019;145:3115-23.
- Lapunzina Badía P, del Campo Casanelles M, Delicado Navarro A, Fernández Toral A, García Alix l, García Guereta LA, et al. Guía para el síndrome de Beckwith-Wiedemann. An Pediatr (Barc). 2006;64:252-9.
- Butti N, Castagna A, Montirosso R. Psychosocial Difficulties in Preschool-Age Children with Beckwith–Wiedemann Syndrome: An Exploratory Study. Children. 2022;9:551.
- Gazzin A, Carli D, Sirchia F, Molinatto C, Cardaropoli S, Palumbo G, et al. Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. Am J Med Genet. 2019;179:1691-1702.
- Bermúdez de Castro López E, Díaz Cirujano AI, Madrigal Terrazas AI, Agud Berenguer C, Demchuk l. El papel del pediatra ante una enfermedad rara. Síndrome de Beckwith Wiedemann. Rev Pediatr Aten Primaria. 2014;16:e111-e114.