
Vol. 24 - Num. 95
Notas clínicas
Cistinuria como causa de litiasis urinaria
Florencia Medina Jereza, Marta Castillo Rodenasb
aMedicina familiar y comunitaria. Área Básica de Salud BS Manresa 4. Manresa. Barcelona. España.
bPediatra. Área Básica de Salud Cardona. Barcelona. España.
Correspondencia: F Medina. Correo electrónico: fmedina.cc.ics@gencat.cat
Cómo citar este artículo: Medina Jerez F, Castillo Rodenas M. Cistinuria como causa de litiasis urinaria . Rev Pediatr Aten Primaria. 2022;24:301-3.
Publicado en Internet: 11-10-2022 - Número de visitas: 12726
Resumen
La cistinuria es una enfermedad genética que provoca un defecto de reabsorción de cistina, causando como manifestación principal litiasis urinarias que pueden llegar a ser de gran tamaño. Es importante tratarla desde temprana edad porque puede comportar importantes comorbilidades.
Palabras clave
● CistinuriaINTRODUCCIÓN
La litiasis urinaria es una entidad infrecuente en Pediatría. Entre las causas genéticas de litiasis, la cistinuria es una causa poco común que se confirma al evaluar la composición de la litiasis extraída o bien mediante estudio genético (cuando hay un familiar afectado y se detecta el gen alterado).
Es importante tratarla desde edad temprana, ya que de no hacerlo las consecuencias pueden ser graves, como la hipertensión arterial o la insuficiencia renal.
CASO CLÍNICO
Paciente varón de 2 años con antecedentes de prematuridad moderada y gestación gemelar. Padres sanos no consanguíneos. Tres hermanos, dos mayores sanos, un gemelo también sin patología conocida.
Consulta a urgencias de nuestro centro de Atención Primaria por clínica de disuria sin fiebre. Se realiza una tira de orina donde presenta leucocituria y hematuria sin nitritos, por lo que se orienta como una cistitis hemorrágica.
Se pide urocultivo, que es negativo. Dada la edad del paciente, se realiza una ecografía renal donde se observa una litiasis piélica de 2 cm con dilatación retrógrada infundibulo-calicilar derecha y otra litiasis de 4 mm en el mismo riñón.
Se realiza estudio de calcio/creatinina que resulta normal, por lo que se descarta hipercalciuria idiopática.
El paciente es derivado a Cirugía pediátrica, donde se le pide una tomografía computarizada (TC) de abdomen para corroborar el hallazgo. En este se observa litiasis pseudocoraliforme en la pelvis renal derecha de 37 mm de diámetro longitudinal con ectasia pielocalicial y múltiples litiasis en grupos caliciales medio e inferior derechos (Figura 1).
| Figura 1. Litiasis urinaria por cistinuria. Imagen de TC del paciente, donde se observa litiasis renal derecha coraliforme |
|---|
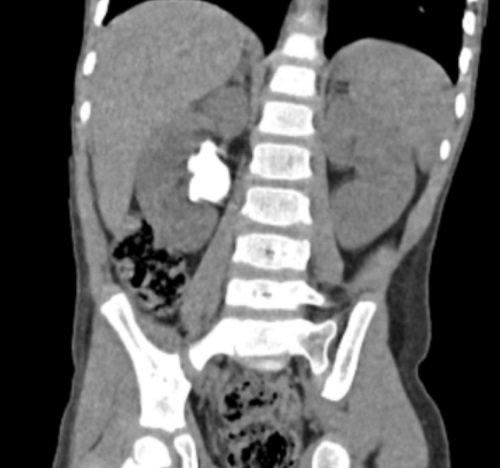 |

Se practica gammagrafía renal con ácido dimercaptosuccínico (DMSA) que informa de un riñón derecho normofuncionante, ligeramente aumentado de tamaño respecto al contralateral y con dilataciones pielocaliciales, sin evidencia de lesiones parenquimatosas y un riñón izquierdo normal con una función renal diferencial 46-54% (valor normal 50 +/-5%).
Se decide realizar una nefrolitotomía percutánea con extracción parcial de la litiasis más grande, que se analiza, pudiendo diagnosticar la cistinuria. Se le deja un catéter para extraer el resto de los cálculos en una segunda intervención (Figura 2).
| Figura 2. Litiasis urinaria por cistinuria. Cálculo renal fragmentado extraído en la intervención. Peso: 1,28 g |
|---|
 |

También se realiza estudio genético, en el cual se encuentra que el paciente está afectado de dos variantes de secuencia en heterocigosis en el gen SLC7A9, característico de la cistinuria. Los padres son heterocigotos para una de las variantes SLC7A9 y tienen una probabilidad del 25% de tener otro hijo afectado por cistinuria en cada gestación. Todos los hijos del paciente serán portadores heterocigotos de una variante en el gen SLC7A9. Se recomienda asesoramiento genético. Los tres hermanos del paciente, uno de los cuales es gemelo, se encuentran pendientes del estudio genético. A los tres se les ha realizado ecografía renal, sin detectarse litiasis urinarias.
Como tratamiento se propone citrato sódico y potásico en solución oral, aparte de una dieta baja en metionina y sodio, ingesta alta de líquidos y rica en residuo alcalino para conseguir un pH urinario alrededor de 7,5.
DISCUSIÓN
La litiasis renal se define como la presencia de cálculos en el tracto urinario y es una entidad infrecuente en la infancia. Se trata de una patología con alto grado de morbilidad, que puede causar lesiones estructurales en el riñón y en las vías urinarias en edades muy tempranas1.
La formación de litiasis requiere la sobresaturación de ciertos iones en la orina. Los factores más implicados son el volumen total de orina, la acumulación de sustancias favorecedoras de cristalización (calcio, cistina, oxalato, ácido úrico), la acumulación de sustancias inhibidoras de la cristalización y el pH urinario. La causa metabólica más frecuente en la infancia es la hipercalciuria idiopática2.
En nuestro caso, después de analizarse la litiasis, se diagnosticó al paciente de cistinuria, una enfermedad infrecuente que afecta a 1/7000 nacidos vivos3. Se trata de una enfermedad con herencia autosómica recesiva o dominante, con una gran variedad de mutaciones diferentes, que produce un defecto en la absorción de cistina y de aminoácidos dibásicos (lisina, arginina y ornitina). Este exceso de cistina urinaria y su gran insolubilidad a pH ácido genera la formación de litiasis de cistina3.
Hay una gran variabilidad en la presentación de la cistinuria entre pacientes, incluso en hermanos con la misma mutación3. La manifestación clínica más frecuente de esta enfermedad son las litiasis urinarias, que según la edad dará una clínica diferente. La clínica típica de cólico nefrítico no se suele presentar tan claramente como en el adulto, sólo en niños mayores y adolescentes. En niños más pequeños, la sintomatología del cólico suele ser inespecífica, pudiéndose manifestar como un cuadro de disuria, irritabilidad, dolor inespecífico o cuadro infeccioso. En los de menos edad las litiasis suelen cursar de forma asintomática o con microhematuria, que se diagnostica de forma casual en el seguimiento de una uropatía o en el estudio de dolor abdominal crónico4.
Dentro de esta entidad, es importante destacar el síndrome de cistinuria-hipotonía, causado por una microdeleción de un gen. Este síndrome se caracteriza por hipotonía al nacimiento, retraso en el crecimiento, alteraciones en el desarrollo y nefrolitiasis3,5.
El diagnóstico se realiza habitualmente en la evaluación de una litiasis renal3. La presencia de cristaluria cistínica (cristales de forma hexagonal) al examen microscópico se considera patognomónica de la enfermedad y se suele realizar en casos de sospecha1. La confirmación diagnóstica se puede realizar mediante la detección de niveles elevados de cistina y aminoácidos dibásicos en orina de 24 horas mediante cromatografía de intercambio iónico y/o espectrometría de masas5.
Se recomienda que se haga un estudio genético a los pacientes para confirmar el diagnóstico y ver la alteración genética para aconsejar al paciente y a sus familiares3,5.
El tratamiento consiste en una ingesta elevada de agua, restricción de sodio y alcalinización urinaria (pH >7) mediante citrato sódico y potásico, requiriendo ocasionalmente la asociación de fármacos que solubilizan la cistina, como la penicilamina, a pesar de que su toxicidad limita su utilidad. El captopril tiene mejor perfil de seguridad, pero es menos eficaz3,5.
Es muy importante el control estricto de la respuesta al tratamiento para disminuir las comorbilidades, como la hipertensión arterial y la insuficiencia renal. Es importante controlar la adherencia terapéutica, ya que muchos pacientes dejan el tratamiento3.
CONFLICTO DE INTERESES
Los autores declaran no tener intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
DMSA: gammagrafía renal con ácido dimercaptosuccínico · TC: tomografía computarizada.
BIBLIOGRAFÍA
- Rodrigo Jiménez MD, Vicente Calderón C. Litiasis renal e hipercalciuria idiopática. Protoc Diagn Ter Pediatr. 2014;1:155-70.
- Pérez Candás JI, Ordóñez Alonso MA, García Nieto V. La litiasis renal y la prelitiasis en la edad pediátrica. Form Act Pediatr Aten Prim. 2014;7;119-32.
- Roussaud F, Lopez de Heredia M, García S, Nunes V, Angerri O. Cistinuria. En: Nefrología al día [en línea] [consultado el 25/09/2022]. Disponible en www.nefrologiaaldia.org/es-articulo-cistinuria-272
- Camacho díaz JA, Vila Cots J. Litiasis renal. Protoc Diagn Ter Pediatr. 2017;1:189-96.
- Servais A, Thomas K, Dello Strologo l, Sayer JA, Bekri S, Bertholet-Thomas A, et al. Cystinuria: clinical practice recommendation. Kidney Int. 2021;99:48-58.