
Vol. 23 - Num. 89
Notas clínicas
Hiperplasia endotelial papilar intravascular en la región supraclavicular
Aída M.ª Gutiérrez Sáncheza, Ana Calabuig Adobesa, Gema Carmen Marcéna, Natalia Estrada Mallarinob, Ainara González Esquedac, Carmelo Guerrero Laleonaa
aServicio de Pediatría. Hospital Infantil Universitario Miguel Servet. Zaragoza. España.
bServicio de Anatomía Patológica. Hospital Universitario Miguel Servet. Zaragoza. España.
cServicio de Cirugía Pediátrica. Hospital Universitario Miguel Servet. Zaragoza. España.
Correspondencia: AM Gutiérrez. Correo electrónico: amgsjm@gmail.com
Cómo citar este artículo: Gutiérrez Sánchez AM, Calabuig Adobes A, Carmen Marcén G, Estrada Mallarino N, González Esqueda A, Guerrero Laleona C. Hiperplasia endotelial papilar intravascular en la región supraclavicular. Rev Pediatr Aten Primaria. 2021;23:87-90.
Publicado en Internet: 30-03-2021 - Número de visitas: 11443
Resumen
La hiperplasia endotelial papilar intravascular (IPEH) es una lesión vascular benigna poco frecuente que se presenta habitualmente como una neoformación subcutánea eritemato-violácea inespecífica. El estudio histopatológico, necesario para el diagnóstico de confirmación, muestra proliferación papilar de células endoteliales asociada con material trombótico. La IPEH puede simular otras lesiones como el angiosarcoma, por lo que el diagnóstico correcto de esta entidad es esencial para evitar tratamientos agresivos. La resección con márgenes amplios suele ser suficiente.
Palabras clave
● Hiperplasia endotelial papilar intravascular ● Lesión vascular benigna ● Tumor de MassonINTRODUCCIÓN
La hiperplasia endotelial papilar intravascular (IPEH), también conocida como tumor de Masson, es una lesión vascular benigna poco frecuente que se localiza habitualmente en extremidades superiores, cabeza y cuello, aunque se han descrito casos en múltiples localizaciones del cuerpo humano. No presenta una morfología característica y se identifican tres variantes: pura, mixta y extravascular1-3. Se manifiesta generalmente como una neoformación subcutánea eritemato-violácea, de crecimiento lento y progresivo, pudiendo simular otras lesiones benignas o malignas como el angiosarcoma. Histológicamente, se caracteriza por proliferación papilar de células endoteliales, asociada con material trombótico.
CASO CLÍNICO
Niño de nueve años, sin antecedentes personales de interés, que presenta una tumoración en región supraclavicular derecha de dos semanas de evolución. Afebril, sin otra clínica acompañante. No refieren cuadro infeccioso los días previos ni antecedente traumático. No viajes recientes, no contacto con animales ni ambiente epidémico familiar.
A la exploración física presenta una tumoración supraclavicular derecha de 2 × 1 cm, de morfología ovalada, blanda, bien delimitada, no adherida a planos profundos y sin signos inflamatorios locales. Es desplazable con la respiración, palpándose mejor con la maniobra de Valsalva mantenida. No presenta adenopatías ni tumoraciones palpables a otros niveles. Resto de exploración física normal.
Se solicitan las siguientes pruebas complementarias: analítica de sangre con hemograma (leucocitos 6530/mm3 [neutrófilos 3400/mm3, linfocitos 2500/mm3, monocitos 300/mm3], hemoglobina 13,4 g/dl, plaquetas 299 000/mm3); proteína C reactiva 0,01 mg/dl, procalcitonina 0,04 ng/ml. Serologías (virus de Epstein-Barr, citomegalovirus, Toxoplasma gondii y Bartonella henselae) negativas. Radiografía de tórax sin alteraciones. Ecografía de partes blandas: nódulo subcutáneo de 24 × 9,9 mm en hueco supraclavicular derecho, de morfología ovoidea, bien delimitado, contornos lisos, encapsulado, con contenido heterogéneo con zonas ecogénicas laminares y refuerzo acústico posterior, que sugiere lesión benigna.
Se biopsia la lesión, objetivándose durante la intervención una lesión violácea que se rompe a la manipulación, muy sangrante. Se ligan y disecan vasos perforantes, simulando la tumoración una adenia necrotizada. Finalmente se realiza extirpación completa de la lesión.
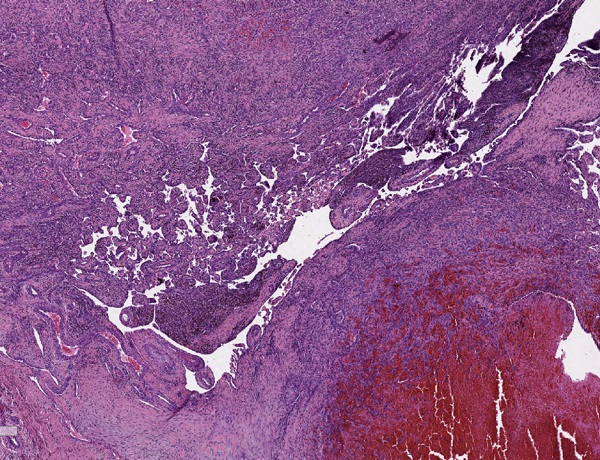
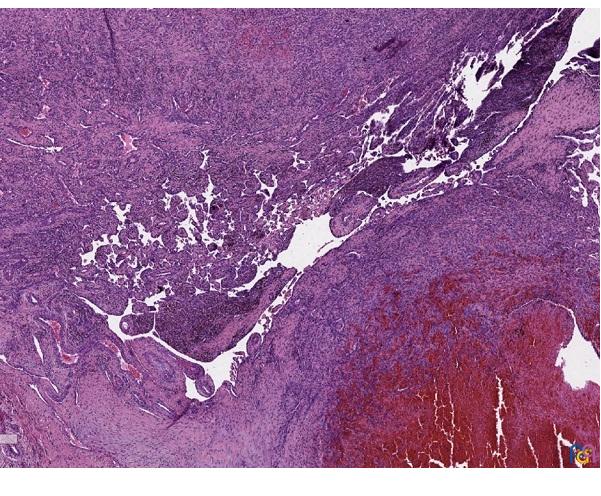
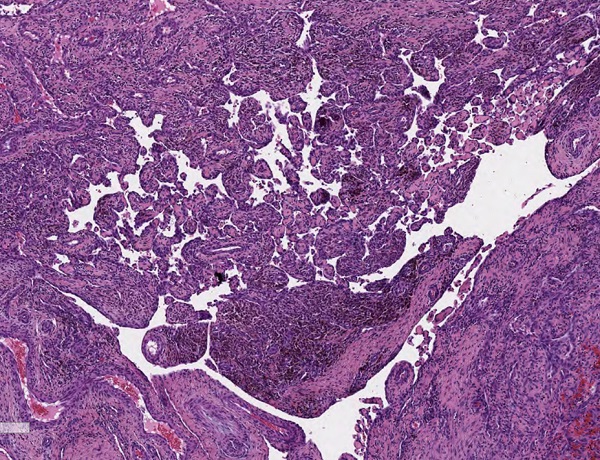
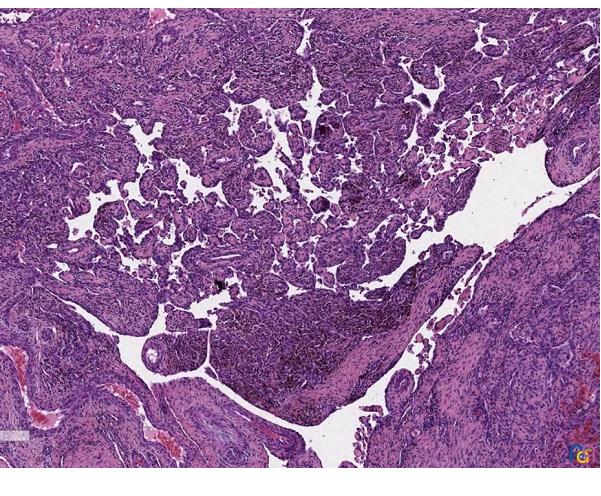
Los resultados de la anatomía patológica confirman el diagnóstico de IPEH. Macroscópicamente se observa una formación nodular de 2,8 × 2 × 1 cm, de coloración parda y superficie irregular. A la seriación, aspecto homogéneo, rojo-violáceo sin necrosis. Microscópicamente se observa una proliferación intravascular de estructuras papilares que se anastomosan entre sí, formando una red. Algunos ejes están compuestos por fibrina y otros por tejido fibroconectivo, encontrándose revestidos por una capa de células endoteliales, sin atipia. La proliferación está en relación con trombo en organización (Figs. 1 y 2).
| Figura 1. Hematoxilina y eosina 25×. Proliferación papilar intravascular, en relación con trombo en organización. |
|---|
 |
| Figura 2. Hematoxicilina y eosina 40×. Proliferación papilar intravascular. |
|---|
 |
Tras la escisión quirúrgica, el paciente presenta una evolución favorable sin observarse recurrencia de la lesión.
DISCUSIÓN
La IPEH es una rara proliferación no neoplásica reactiva endotelial, frecuentemente localizada en la piel y tejidos subcutáneos, a pesar de haberse descrito en otras localizaciones extracutáneas4. Existen pocos casos en edad pediátrica reportados en la literatura médica.
Fue descrita por primera vez en 1923 por Pierre Masson como una neoplasia peculiar en venas hemorroidales, que histológicamente se caracterizaba por una proliferación papilar endotelial atípica que simulaba un angiosarcoma5.
Se considera un tumor de estirpe vascular y comportamiento benigno que no presenta ningún aspecto clínico específico. Puede manifestarse a cualquier edad, con predominio entre la tercera y cuarta décadas de la vida. Es más frecuente en mujeres6 y representa el 2-4% de todas las proliferaciones vasculares de piel y mucosas7.
Existen clásicamente 3 tipos de IPEH según la clasificación de Hashimoto1,2: forma pura o primaria, la más frecuente, que se desarrolla en espacios vasculares dilatados y afecta a las extremidades, la cabeza y el cuello; forma mixta o reactiva secundaria, que suele ser intramuscular y ocurre en anomalías vasculares ya existentes como hemangiomas, varices, malformaciones arteriovenosas y granuloma piógeno; y la forma extravascular, variedad excepcional asociada con la formación de hematomas inducidos por traumatismo.
El diagnóstico de sospecha es clínico, con confirmación anatomopatológica. A nivel histológico, la lesión principal consiste en un entramado de conductos vasculares anastomosados, con proyecciones intraluminales. Los vasos se muestran de carácter infiltrativo, con células endoteliales hiperplásicas, algunas hipercromáticas, anastomosadas e incluso con algunos núcleos pleomórficos, que puede llegar a confundirse con un proceso sarcomatoso. Es frecuente encontrar eritrocitos en la luz vascular. El estroma está conformado por un material eosinofílico y hialino que puede fusionarse con vasos trombosados7.
Es importante realizar un adecuado diagnóstico diferencial con otras entidades, para evitar tratamientos inapropiados. Algunas características que pueden ayudar a distinguir la IPEH del angiosarcoma son la circunscripción de la lesión, la localización en un vaso o su asociación con trombosis y la arquitectura papilar sin atipia citológica significativa o áreas de crecimiento sólido8.
El tratamiento consiste en la resección completa de la tumoración, incluyendo márgenes amplios para evitar su recurrencia. En las formas puras, bien circunscritas y con límites evidentes, una escisión con 2 mm de margen es suficiente, presentando bajas tasas de recurrencia. En las formas mixtas que asientan sobre anomalías vasculares subyacentes, se pueden requerir márgenes más amplios para evitar la recurrencia de la lesión3,9,10.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
IPEH: hiperplasia endotelial papilar intravascular.
BIBLIOGRAFÍA
- Hashimoto H, Daimaru Y, Enjoji M. Intravascular papillary endothelial hyperplasia. A clinicopathologic study of 91 cases. Am J Dermatopathol. 1983;5:539-46.
- Higashi Y, Uchida Y, Yoshii N, Kubo H, Kanzaki T, Yokouchi M, et al. Multiple intravascular papillary endothelial hyperplasia affecting skin and bone. Clin Exp Dermatol. 2009;34:740-3.
- Anderson JC, Brown KK. Masson tumor arising in a congenital vascular anomaly. Pediatr Dermatol. 2013;30:745-7.
- Mestiri S, Karoui M, Charachon A, Deux JF, Chaumette MT. Intravascular papillary endothelial hyperplasia of the jejunum: An unusual cause of melena. Int J Surg Pathol. 2007;15:192-5.
- Masson P. Hèmangioendothèliome vègètant intravasculaire. Bull Soc Anat Paris. 1923;93:517-23.
- Schwartz SA, Taljanovic MS, Harrigal CL, Graham AR, Smyth SH. Intravascular papillary endothelial hyperplasia: sonographic appearance with histopathologic correlation. J Ultrasound Med. 2008;27:1651-3.
- Cardona-Hernández MA, Fierro-Arias R, González-González M, Cabrera-Pérez AL. Hiperplasia endotelial papilar intravascular (tumor de Masson). Dermatol Rev Mex. 2017;61:398-403.
- Fernández García-Guilarte R, Enríquez de Salamanca Celada J, Comenero I. Hiperplasia papilar endotelial intravascular. Cir Plást Iberolatinoam. 2009;35:155-8.
- Paunipagar BK, Rasalkar DD, Ng A, Griffith JF, Bagaria V. Intravascular papillary endothelial hyperplasia: Report of two cases. Acta Radiol. 2011;52:499-502.
- Juan YH, Huang GS, Chiu YC, Chang WC, Hsu YC. Intravascular papillary endothelial hyperplasia of the calf in an infant: MR features with histological correlation. Pediatr Radiol. 2009;39:282-5.