
Vol. 21 - Num. 81
Originales
Hipertensión intracraneal idiopática: casuística y revisión de la bibliografía
María Álvarez Casañoa, Rocío Calvo Medinab, Jacinto Martínez Antónb
aPediatra. CS Alcolea. Córdoba. España .
bNeuropediatría. Servicio de Pediatría. Hospital Regional Universitario de Málaga. Málaga. España.
Correspondencia: M Álvarez . Correo electrónico: mariaac22@hotmail.com
Cómo citar este artículo: Álvarez Casaño M, Calvo Medina R, Martínez Antón J. Hipertensión intracraneal idiopática: casuística y revisión de la bibliografía. Rev Pediatr Aten Primaria. 2019;21:15-20.
Publicado en Internet: 13-02-2019 - Número de visitas: 37306
Resumen
Introducción: se analizan las características de la hipertensión intracraneal idiopática; clínica al inicio, pruebas realizadas, tratamiento y evolución que presentaron.
Material y métodos: estudio descriptivo retrospectivo de los pacientes diagnosticados de hipertensión intracraneal idiopática en los últimos siete años (2011-2017) en un hospital de tercer nivel.
Resultados: se estudiaron 40 pacientes (55% mujeres) con una edad media de 9,6 años. Como factores asociados, el 5% seguían tratamiento con hormona del crecimiento. Solo el 25% presentaban sobrepeso u obesidad. El síntoma principal fue cefalea opresiva, asociando vómitos (27,5%) o alteraciones visuales (22%). Tres pacientes presentaron hallazgo casual de papiledema bilateral. La exploración física fue anodina (65%), se observó estrabismo por parálisis del VI par craneal (35%). Presentaron papiledema el 62,5%. La campimetría solo se realizó en el 55% de los pacientes y estaba alterada en el 50% de estos. El tiempo hasta el diagnóstico fue 44,8 días. Obtuvimos una presión de apertura media 29,7 cm H2O (± 8,2). Se realizó tomografía computarizada al 85% de los pacientes y fue normal en el 88,2% de estos. Se hizo resonancia magnética craneal al 7,5%, y fue normal en el 70% de estos. Se practico angio-RM al 5%, y fue normal en todos los casos. Se solicitó analítica, con función renal y hepática (62,5%, todos normal), hormonal (65%), estudio de trombofilias y autoinmunidad (10% y 20% respectivamente, anodinas). Se inició tratamiento con acetazolamida (95%), y hubo que añadir corticoterapia por falta de respuesta en el 24% de los casos. En el 5% se autolimitó espontáneamente. Como último escalón, el 7,5% requirió válvula de derivación lumboperitoneal. Evolucionaron favorablemente el 95%, con recidivas en el 15% de los casos. El tiempo medio hasta la resolución fue de 3,9 meses.
Conclusión: el diagnóstico y tratamiento precoz de la hipertensión intracraneal idiopática es importante para evitar posibles secuelas irreversibles. El estudio oftalmológico, especialmente la campimetría, es esencial para el diagnóstico, seguimiento y determinación de la agresividad del tratamiento.
Palabras clave
● Campimetría ● Hipertensión intracraneal ● PapiledemaINTRODUCCIÓN
La hipertensión intracraneal idiopática (HII) o hipertensión intracraneal benigna es una enfermedad poco común en la infancia. Se define por una serie de signos y síntomas producidos por el aumento de la presión intracraneal (cefalea, papiledema y afectación visual), y es necesario realizar un diagnóstico de exclusión de otras posibles causas de hipertensión intracraneal (lesiones ocupantes de espacio, traumatismos craneoencefálicos, encefalitis o meningitis)1,2. Por lo general, la evolución es benigna, pero dada la posibilidad de un deterioro grave de la visión se recomienda un control estricto de estos pacientes hasta la resolución completa del cuadro.
El objetivo del estudio es realizar un análisis sobre la incidencia de dicha patología en nuestro medio, así como los datos demográficos, clínica al inicio, pruebas realizadas hasta el diagnóstico, tratamiento y evolución que presentaron.
MATERIAL Y MÉTODOS
Se trata de un estudio descriptivo retrospectivo de los pacientes diagnosticados de HII en los últimos siete años (2011-2017) en la consulta de Neuropediatría de un hospital de tercer nivel.
Para el diagnóstico de HII se ha seguido los criterios de Dandy-Smith modificados3 (Tabla 1): con sintomatología clínica compatible, un análisis de líquido cefalorraquídeo (LCR) normal, una presión de apertura de LCR mayor de 20 cm H2O y una prueba de neuroimagen sin hallazgos. Como signos de hipertensión intracraneal, el abombamiento de la fontanela en los lactantes y el papiledema en los niños mayores, asociados o no a otros síntomas, como vómitos, cefalea o parálisis del VI par craneal.
| Tabla 1. Criterios de Dandy-Smith modificados3 |
|---|
| Signos y síntomas de aumento de presión intracraneal, incluido el papiledema |
| Ausencia de focalidad neurológica, a excepción de paresia del VI par craneal |
| Neuroimagen normal (se admite la existencia de ventrículos pequeños e imagen de silla turca vacía) |
| Aumento de la presión de apertura de líquido cefalorraquídeo (mayor de 200-250 mm de agua), pero de composición normal |
| Paciente consciente y alerta |
| Sin causa alternativa que explique el aumento de la presión intracraneal |

Se consultaron los datos en las historias clínicas de los pacientes dados de alta con el diagnóstico de HII en la consulta de neuropediatría en el periodo comprendido entre enero de 2011 y diciembre de 2017. Todos ellos habían aceptado el consentimiento para la realización de la punción lumbar y estudio de los resultados.
RESULTADOS
Se obtuvo una muestra de 40 pacientes, una vez excluidos a los que no se llegó al diagnóstico definitivo de HII.
El 55% eran mujeres, la edad media de la muestra fue de 9,6 ± 3,04 años. El 47,5% de los pacientes acudió en primer lugar a Urgencias, mientras que un 20% fue a la consulta de Neuropediatría derivados desde Atención Primaria, y otro 20% consultó directamente en Oftalmología.
No se observó mayor prevalencia en ninguna estación del año. El 93% refería un desarrollo psicomotor adecuado. Entre los antecedentes personales, el 47,5% no tenía antecedentes de interés, el 15% presentaba defectos de refracción y el 15% algún antecedente neurológico (migraña, trastornos del desarrollo o epilepsia en uno de los casos). El 25% de los pacientes tenía antecedentes familiares de migraña. Como factores asociados, el 5% estaban en tratamiento con hormona del crecimiento (GH) por talla baja. El 17,5% refería una infección previa (sinusitis, faringitis u otitis media aguda), y traumatismo craneoencefálico previo tan solo el 2,5%. Un dato que hay que remarcar es que tan solo el 25% presentaban sobrepeso u obesidad.
El síntoma principal presente en el 92,7% de los pacientes fue la cefalea, de características opresiva y holocraneal, sin predominio horario, el 27,5% asoció vómitos y el 22% alteraciones de la visión (diplopia o visión borrosa). Uno de ellos asociaba episodios paroxísticos. Tres pacientes no presentaron ninguna sintomatología, fueron derivados ante el hallazgo casual de papiledema bilateral.
La exploración física fue anodina en el 65%, observándose un estrabismo por parálisis del VI par craneal en el 35% de los pacientes. En la exploración oftalmológica presentaron papiledema el 62,5%, y la campimetría tan solo se realizó en el 55% de los pacientes, y estaba alterada en el 50% de estos.
El tiempo de evolución desde el inicio de la sintomatología hasta el diagnóstico fue de 44,8 días (61,3). La HII es una patología en la que es necesario realizar un diagnóstico de exclusión, por lo que en el 100% de los pacientes se realizó punción lumbar y prueba de neuroimagen.
Se consideró patológico un valor de la presión de apertura del LCR mayor a 20 cm H2O; obtuvimos en los pacientes una media de 29,7 cm H2O (±8,2).
La citología del LCR fue normal en el 100% de los casos. Como prueba de imagen se realizó tomografía computarizada (TC) en el 85% de los pacientes, con resultado normal en el 88,2% de ellos, y con hallazgos inespecíficos en el resto (dos casos presentaban un quiste aracnoideo de pequeño tamaño sin efecto masa, un caso sinusitis y en otro caso se observaron calcificaciones supratentoriales extraaxiales). Se realizó resonancia magnética (RM) de cráneo en el 87,5% de los casos, con resultado normal en el 70% de estos; un caso presentó una malformación de los senos venosos (2,8%), el 8,5% sinusitis, dos pacientes un quiste aracnoideo de pequeño tamaño (5,7%), y hallazgos inespecíficos, como aumento de señal cortical en el lóbulo occipital bilateral en un caso (2,8%), aplanamiento de la glándula hipofisaria en otro paciente (2,8%), aumento del tamaño de las astas occipitales y temporales en otro (2,8%) y en el paciente con antecedentes de epilepsia y calcificaciones en el TC, se observó en la RM una zona de sangrado evolucionado en la cápsula externa izquierda del lóbulo temporal (2,8%). Se realizó angio-RM en el 5% de los casos, todos ellos con resultado normal, salvo la malformación de los senos venosos.
La electroencefalografía (EEG) se realizó en el 5% de los casos (el paciente que presentaba epilepsia de base y aquel que asociaba episodios paroxísticos además de la cefalea).
En función de los antecedentes personales, familiares y el desarrollo puberal, se solicitaron determinaciones analíticas, como el estudio de la función renal y hepática (62,5%, todos normales), determinaciones hormonales (65%, se detectaron anomalías en el 7,6%: hiperandrogenismo e hipotiroidismo), estudio de trombofilias y estudio de autoinmunidad (10 y 20% respectivamente, sin detectarse anomalías).
El tratamiento recibido en el 95% de los casos fue médico, con acetazolamida, un inhibidor de la anhidrasa carbónica, en el 24% de los casos hubo que añadir corticoterapia por falta de respuesta. En el 5% restante, la clínica se autolimitó espontáneamente. El corticoide añadido en los que lo precisaron fue la prednisona en el 18,5% y dexametasona en el 5%. Como último escalón terapéutico, requirieron la colocación de una válvula de derivación lumboperitoneal el 7,5% de los pacientes.
El 95% de los pacientes tuvo respuesta favorable al tratamiento médico; el 15% presentó recidiva de la HII. Remarcar que el tiempo medio de evolución hasta la resolución de los síntomas y normalización del fondo de ojo fue de 3,9 meses (rango: 1-18 meses).
En todos los pacientes, la resolución del proceso fue completa, con desaparición del papiledema y sin precisar tratamiento de mantenimiento, aunque sí se observaron recurrencias en seis de los casos.
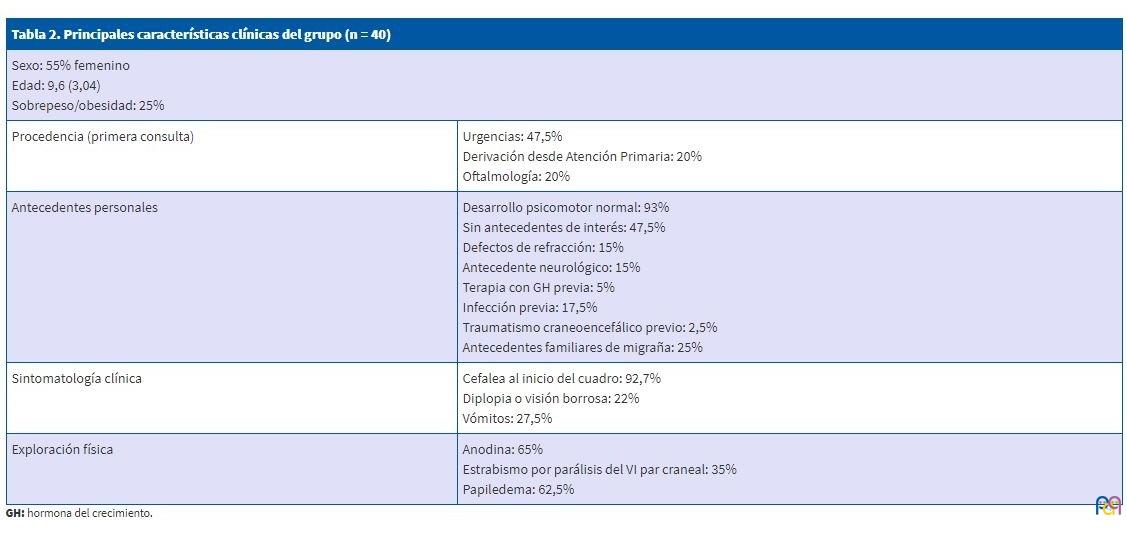
En las Tablas 2 y 3 se recogen los principales datos clínicos de nuestro estudio.
| Tabla 2. Principales características clínicas del grupo (n = 40) | |
|---|---|
| Sexo: 55% femenino Edad: 9,6 (3,04) años Sobrepeso/obesidad: 25% |
|
| Procedencia (primera consulta) | Urgencias: 47,5% Derivación desde Atención Primaria: 20% Oftalmología: 20% |
| Antecedentes personales | Desarrollo psicomotor normal: 93% Sin antecedentes de interés: 47,5% Defectos de refracción: 15% Antecedente neurológico: 15% Terapia con GH previa: 5% Infección previa: 17,5% Traumatismo craneoencefálico previo: 2,5% Antecedentes familiares de migraña: 25% |
| Sintomatología clínica | Cefalea al inicio del cuadro: 92,7% Diplopia o visión borrosa: 22% Vómitos: 27,5% |
| Exploración física | Anodina: 65% Estrabismo por parálisis del VI par craneal: 35% Papiledema: 62,5% |
| Tabla 3. Pruebas diagnósticas, tratamiento y evolución de los pacientes | |
|---|---|
| Pruebas complementarias realizadas | Campimetría: 18/40 (45%). Normal 9/18 (50%). Alterada 9/18 (50%) Presión de apertura del LCR: 29,7 cm H2O Realización de TC: 85% Realización de RM craneal: 87,8% Realización de angio-RMN: 5% Realización de EEG: 5% Función renal y hepática: 62,5% Estudio hormonal: 65% Estudio de trombofilias: 10% Estudio de autoinmunidad: 20% |
| Tratamiento | Resolución espontánea (retirada del factor desencadenante): 5% Acetazolamida: 95% Acetazolamida + corticoterapia: 24% DLP: 7,5% Tiempo medio hasta resolución: 3,9 meses (rango: 1-18 meses) Recidiva: 15% |

DISCUSIÓN
La HII es una patología poco frecuente y con potenciales riesgos visuales en los niños. En la población adulta se describe una incidencia de 1-2 por 100 000, y es mayor su incidencia en mujeres obesas de 15 a 44 años2. En la población pediátrica no existen datos de incidencia específicos, aunque también se describe principalmente en niñas adolescentes con sobrepeso, pudiendo ser el único factor desencadenante. Sin embargo, en nuestra muestra no hemos podido demostrarlo puesto que no existe una mayor incidencia significativa en niñas (55%) y tan solo presentaron sobrepeso el 25% de los pacientes.
Actualmente es una entidad de etiología desconocida, aunque hay varias teorías sobre su origen. Se ha descrito una asociación directa con diferentes fármacos como posibles desencadenantes, como los corticoides, la GH (presente en el 5% de nuestra muestra), hipervitaminosis A, tetraciclinas o la existencia de una malformación de los senos venosos (presente en un paciente de nuestra serie)3,4.
El motivo de consulta más frecuente es la cefalea asociada a vómitos o a alteraciones visuales (visión borrosa por edema de papila o diplopía por parálisis del VI par craneal). En nuestro estudio, la cefalea de tipo opresiva holocraneal es el síntoma cardinal, y se asocia a la afectación visual en el 22% de los casos. Una minoría de la muestra presentó papiledema bilateral como hallazgo casual en una revisión oftalmológica rutinaria, sin tener ninguna otra sintomatología.
La HII es un diagnóstico de exclusión y deben descartarse otras causas de hipertensión intracraneal, como encefalitis o meningitis, trombosis venosas, o mucho menos frecuentes carcinomatosis leptomeníngeas o melanosis neurocutáneas, entre otras.
El tratamiento se instaura para lograr la remisión de los síntomas y preservar la función visual, evitando posibles secuelas al disminuir la presión intracraneal.
En nuestra muestra, en los dos pacientes en los que la HII se asociaba al tratamiento con GH, este se suspendió con resolución de la clínica. En el resto se instauró tratamiento médico con acetazolamida como primera línea, en dosis de 25-30 mg/kg/día repartidas en dos tomas diarias. La acetazolamida es un inhibidor de la anhidrasa carbónica que actúa disminuyendo la producción de LCR, aunque para ello se requieren dosis altas, que pueden llegar a ser mal toleradas, en cuyo caso se puede sustituir por furosemida o asociar un corticoide. El uso de corticoides puede ser útil como tratamiento adyuvante de rescate en pacientes con rápido deterioro de la función visual mientras se plantea la posibilidad de cirugía. Es importante hacer una retirada escalonada y progresiva de los corticoides, puesto que si se realiza de manera brusca puede llevar a un aumento de la presión intracraneal por rebote.
En la población adulta se emplean otras líneas de tratamiento, como el topiramato y la zonisamida, también inhibidores de la anhidrasa carbónica. Sin embargo, no existen evidencias de su uso en Pediatría.
Como última línea de tratamiento, se emplea la cirugía en casos de deterioro progresivo de la función visual o cefalea rebelde a pesar del tratamiento médico. Entre los procedimientos invasivos están la realización de punciones lumbares repetidas o la derivación lumboperitoneal o ventriculoperitoneal, u otras más encaminadas a tratar el papiledema, como la descompresión o fenestración de la vaina del nervio óptico (aunque la experiencia es limitada en niños).
Tras revisar la literatura médica actual, no se han descrito nuevas líneas de tratamiento. La realización de punciones evacuadoras seriadas ha disminuido de forma marcada, por tratarse de un tratamiento cruento5-7.
A pesar de que en la literatura médica se describe una alta tasa de resolución espontánea, nosotros preferimos instaurar el tratamiento médico de forma precoz por el riesgo de afectación visual1.
Es importante realizar controles clínicos y oftalmológicos periódicos. No solo del fondo de ojo, sino que es fundamental realizar campimetrías seriadas por el oftalmólogo, puesto que su afectación indica empeoramiento y avance de la enfermedad.
En el momento en que la exploración oftalmológica se normaliza hay que retirar de forma progresiva el tratamiento farmacológico. Se aconseja disminuir la acetazolamida en un mínimo de dos meses. Y continuar el seguimiento de los pacientes por la posibilidad de recaídas (8-38% de los pacientes): en nuestra muestra se describió una recidiva en el 15%.
CONCLUSIÓN
La HII es una enfermedad poco frecuente, pero cuyo diagnóstico y tratamiento precoz es esencial para evitar posibles secuelas irreversibles. Es fundamental contar con un equipo multidisciplinar coordinado y compuesto por neuropediatras, oftalmólogos y neurocirujanos.
Hay que hacer hincapié en que el estudio oftalmológico, con la valoración del fondo de ojo y especialmente la campimetría, esencial para el diagnóstico, el seguimiento y la determinación de la agresividad del tratamiento.
Igualmente es importante contar con un protocolo diagnóstico-terapéutico en la unidad, para orientar las pruebas complementarias y decidir el manejo terapéutico que se va a llevar a cabo en estos pacientes.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
EEG: electroencefalografía · GH: hormona del crecimiento · HII: hipertensión intracraneal idiopática · LCR: líquido cefalorraquídeo · RM: resonancia magnética · TC: tomografía computarizada.
BIBLIOGRAFÍA
- Monge Galindo L, Pérez Delgado R, López-Pisón J, Olloqui-Escalona A, García Íñiguez JP, Ruiz del Olmo Izuzquiza L. Hipertensión intracraneal benigna. Experiencia en 18 años. An Pediatr (Barc). 2009;71:400-6.
- Mosquera Gorostidi A, Iridoy Zulet M, Azcona Ganuza G, Gembero Esarte E. Seudotumor cerebri en niños: etiología, características clínicas y evolución. Neurologia. 2017. pii: S0213-4853(16)30244-4.
- Eldes NH, Yilmaz Y. Pseudotumour cerebri in children: etiological, clinical features and treatment modalities. Eur J Paediatr Neurol. 2012;16:349-55.
- Ray WZ, Lee A, Blackburn SL, Lueder GT, Leonard J R. Pseudotumor cerebri following tapered corticosteroid treatment in an 8-month-old infant. J Neurosurg Pediatr. 2008;1:88-90.
- Rodríguez de Rivera FJ, Martínez-Sánchez P, Ojeda-Ruiz de Luna J, Arpa-Gutiérrez FJ, Barreiro-Tella P. Benign intracranial hypertension. History, clinical features and treatment in a series of 41 patients. Rev Neurol. 2003;37:801-5.
- Aylward SC, Way AL. Pediatric intracranial hypertension: a current literature review. Curr Pain Headache Rep. 2018;22:14.
- Youroukos S, Psychou F, Fryssiras S, Paikos P, Nicolaidou P. Idiopathic intracranial hypertension in children. J Child Neurol. 2000;15:453-7.