

Vol. 21 - Num. 81
Original Papers
Idiopathic intracranial hypertension: epidemiology and current literature review
María Álvarez Casañoa, Rocío Calvo Medinab, Jacinto Martínez Antónb
aPediatra. CS Alcolea. Córdoba. España .
bNeuropediatría. Servicio de Pediatría. Hospital Regional Universitario de Málaga. Málaga. España.
Correspondence: M Álvarez . E-mail: mariaac22@hotmail.com
Reference of this article: Álvarez Casaño M, Calvo Medina R, Martínez Antón J. Idiopathic intracranial hypertension: epidemiology and current literature review. Rev Pediatr Aten Primaria. 2019;21:15-20.
Published in Internet: 13-02-2019 - Visits: 24543
Abstract
Introduction: the characteristics of idiopathic intracranial hypertension are analyzed; epidemiology, clinic at the beginning, tests performed, treatment and evolution that presented the analyzed cases.
Material and methods: a retrospective descriptive study was conducted on patients with idiopathic intracranial hypertension in the last seven years (2011-2017), in a third level hospital.
Results: forty patients (55% women) were studied, with a mean age of 9.6 years. As associated factors, 5% had a treatment with growth hormone. It is important to note that only 25% were overweight or obese. The main symptom was oppressive headache, without predominance hours, associating vomiting (27.5%), or visual alterations (22%). Three asymptomatic patients presented a chance finding of bilateral papilledema. The physical examination was anodyne (65%), showing strabismus due to cranial nerve palsy VI (35%). 62,5% presented papilledema, and the campimetry was only performed in 55% of the patients, altered in 50% of them. The time to diagnosis was 44.8 days. We obtained an average opening pressure of 29.7 cm H2O (± 8.2). CT was performed (85%), being normal (88.2%). MRI of the skull (7.5%), normal (70%). Angio-NMR (5%), all normal. Analytical was requested, with renal and hepatic function (62.5%, all normal), hormonal (65%), thrombophilic study and autoimmunity (10% and 20% respectively, anodyne). Treatment was started with acetazolamide (95%), requiring the addition of corticotherapy due to lack of response 24%. In 5%, it spontaneously self-limited. As a last step, 7.5% required a lumboperitoneal bypass valve. They evolved favorably 95%, relapsing 15%. Redialing the average time to resolution was 3.9 months.
Conclusion: idiopathic intracranial hypertension is rare, but its diagnosis and early treatment is essential to avoid possible irreversible sequelae. The ophthalmological study, by fundus and especially campimetry, is essential for the diagnosis, monitoring and determination of the aggressiveness of the treatment.
Keywords
● Intracranial hypertension ● Papilloedema ● Visual field testINTRODUCTIÓN
Idiopathic intracranial hypertension (IIH) or benign intracranial hypertension is an infrequent disease in children. It is characterised by a constellation of signs and symptoms resulting from raised intracranial pressure (headache, papilledema and visual impairment), and its diagnosis requires ruling out other possible causes of intracranial hypertension (space-occupying lesions, head trauma, encephalitis or meningitis).1,2 Its course is usually benign, but given the risk of severe visual impairment, strict monitoring of these patients until full resolution is recommended.
The aim of our study was to analyse the incidence of IIH in our area and the demographic characteristics, tests performed until diagnosis, treatment and outcomes of affected patients.
MATERIALS AND METHODS
We conducted a retrospective descriptive study of the patients that received a diagnosis of IIH in the Department of Paediatric Neurology of a tertiary care hospital in the past 7 years (2011-2017).
We used the modified Dandy-Smith criteria3 to diagnose IIH (Tabla 1): compatible symptoms, normal cerebrospinal fluid (CSF) composition, a CSF opening pressure greater than 20 cm H2O and unremarkable neuroimaging findings. The signs considered indicative of raised intracranial pressure were bulging fontanelle in infants and papilloedema in older children, with or without accompanying symptoms such as vomiting, headache or sixth cranial nerve palsy.
| Table 1. Modified Dandy-Smith criteria3 |
|---|
| Symptoms and signs of increased intracranial pressure, including papilloedema |
| Absence of localizing findings on neurologic examination, with the exception of sixth cranial nerve palsy |
| Normal neuroimaging studies (except for small ventricular size and empty sella turcica) |
| Increased cerebrospinal fluid pressure (> 200-250 mm H2O), with normal fluid composition |
| Patient awake and alert |
| No other cause of increased intracranial pressure present |

We collected data from the electronic health records of patients discharged from the Department of Paediatric Neurology department with a diagnosis of IIH between January 2011 and December 2017. All of them had consented to performance of a lumbar puncture and the analysis of the results.
RESULTS
The final sample consisted of 40 patients after excluding patients without a definitive diagnosis of IIH.
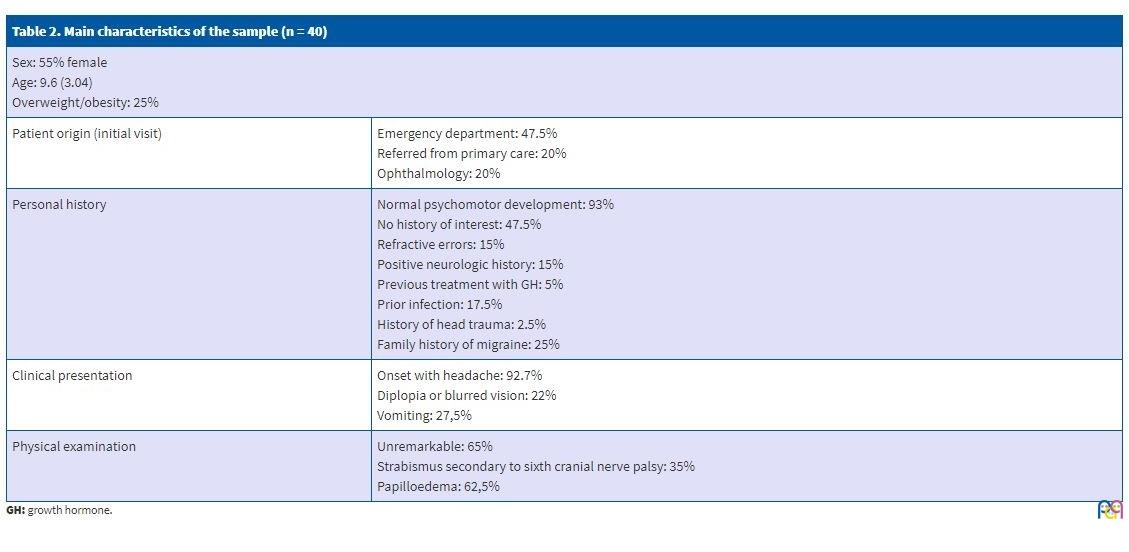
Of all patients, 55% were female, and the mean age of the sample was 9.6 ± 3.04 years. Patients initially sought care in the emergency department in 47.5% of cases, while 20% visited the Department of Paediatric Neurology after being referred from primary care and another 20% sought care directly in the Department of Ophthalmology.
We did not find changes in prevalence based on the season. Of all patients, 93% reported normal psychomotor development. The personal history was unremarkable in 47.5% of patients, while 15% had refractive errors and 15% some form of neurologic problem (migraine, developmental disorders or, in 1 patient, epilepsy). There was a positive family history of migraine in 25% of the patients. When it came to risk factors, 5% were undergoing treatment with growth hormone (GH) due to short stature; 17.5% reported a previous infection (sinusitis, pharyngitis or acute otitis media), and only 2.5% a history of head trauma. We ought to highlight that only 25% of the patients were overweight or obese.
The main presenting symptom, found in 92.7% of patients, was a holocranial tension headache with no changes in intensity based on the time of day, associated with vomiting in 27.5% and with changes in vision (blurred or double vision) in 22%. One patient also experienced paroxysmal events. Three patients were asymptomatic and had been referred for evaluation due to chance finding of bilateral papilloedema.
The physical examination was unremarkable in 65% of patients. Thirty-five percent of patients presented with strabismus secondary to sixth cranial nerve palsy. The ophthalmological evaluation detected papilloedema in 62.5% of patients; a visual field test was performed in only 55% of patients and found abnormalities in half of them.
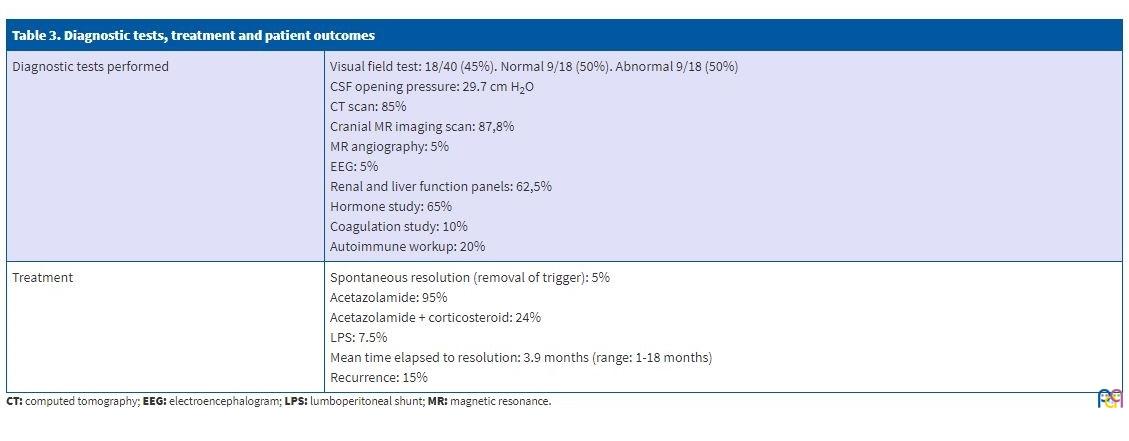
The time elapsed from onset to diagnosis was 44.8 days (61.3). Since IIH is a diagnosis of exclusion, all patients underwent a lumbar puncture and neuroimaging tests.
Values of the CSF opening pressure greater than 20 cm H2O were considered pathological, and the mean pressure found in our patients was 29.7 cm H2O (standard deviation, ±8.2).
The CSF composition was normal in all patients. When it came to imaging tests, 85% of patients underwent a computed tomography (CT) scan, which was normal in 88.2% of them, with nonspecific abnormal findings in the rest (a small arachnoid cyst without mass effect in 2 patients, sinusitis in 1 patient and extra-axial supratentorial calcifications in 1 patient). A cranial magnetic resonance imaging (MRI) scan was performed in 87.5% of patients, with normal findings in 70% of them and detection of a dural sinus malformation in 1 patient (2.8%), of sinusitis in 8.5%, a small arachnoid cyst in 2 patients (5.7%) and nonspecific abnormalities in other patients, including bilateral cortical hyperintensity in the occipital lobe in 1 patient (2.8%), flattening of the pituitary gland in 1 patient (2.8%), enlargement of occipital and temporal horns in 1 patient (2.8%) and, in the patient with a history of epilepsy and calcifications in the CT scan (2.8%), bleeding in the external capsule of the temporal lobe. A magnetic resonance angiogram was performed in 5% of patients, with normal findings except for the dural sinus malformation in 1 patient.
An electroencephalogram (EEG) was performed in 5% of cases (in the patient with underlying epilepsy and the patient with headache associated with paroxysmal events).
Laboratory tests were ordered based on the personal and family history and the stage of pubertal development, such as renal and liver function panels (in 62.5%, normal in all), measurement of hormone levels (in 65%, with abnormalities detected in 7.6%: hyperandrogenism and hypothyroidism), coagulation panel (in 10%, normal in all) and autoimmunity workup (in 20% normal in all).
In 95% of patients, treatment was pharmacological and consisted of acetazolamide, a carbonic anhydrase inhibitor; 24% required the addition of corticosteroids due to nonresponse. In the remaining 5%, the disease resolved spontaneously. In those that required corticosteroid therapy, the drugs given were prednisone (18.5%) and dexamethasone (5%). As a last-tier option, 7.5% of patients required placement of a lumboperitoneal shunt.
Ninety-five percent of patients responded favourably to treatment, and 15% experienced recurrence of IIH. We ought to mention that the mean time elapsed to resolution of symptoms and normalization of the eye fundus was 3.9 months (range: 1-18 months).
In all patients, IIH resolved completely with full regression of the papilloedema and without need of maintenance treatment, although 6 patients experienced recurrences.
Tables 2 and 3 list the main clinical features of the cases included in the study.
| Table 2. Main characteristics of the sample (n = 40) | |
|---|---|
| Sex: 55% female Age: 9.6 (3.04) Overweight/obesity: 25% |
|
| Patient origin (initial visit) | Emergency department: 47.5% Referred from primary care: 20% Ophthalmology: 20% |
| Personal history | Normal psychomotor development: 93% No history of interest: 47.5% Refractive errors: 15% Positive neurologic history: 15% Previous treatment with GH: 5% Prior infection: 17.5% History of head trauma: 2.5% Family history of migraine: 25% |
| Clinical presentation | Onset with headache: 92.7% Diplopia or blurred vision: 22% Vomiting: 27,5% |
| Physical examination | Unremarkable: 65% Strabismus secondary to sixth cranial nerve palsy: 35% Papilloedema: 62,5% |
| Table 3. Diagnostic tests, treatment and patient outcomes | |
|---|---|
| Diagnostic tests performed | Visual field test: 18/40 (45%). Normal 9/18 (50%). Abnormal 9/18 (50%) CSF opening pressure: 29.7 cm H2O CT scan: 85% Cranial MR imaging scan: 87,8% MR angiography: 5% EEG: 5% Renal and liver function panels: 62,5% Hormone study: 65% Coagulation study: 10% Autoimmune workup: 20% |
| Treatment | Spontaneous resolution (removal of trigger): 5% Acetazolamide: 95% Acetazolamide + corticosteroid: 24% LPS: 7.5% Mean time elapsed to resolution: 3.9 months (range: 1-18 months) Recurrence: 15% |
DISCUSSION
Idiopathic intracranial hypertension is a rare disease that may cause visual impairment in children. The incidence reported in adults is of 1 to 2 per 100 000 individuals, and is higher in obese women aged 15 to 44 years.2 There are no specific data on the incidence of IIH in the paediatric population, although it also has been mainly described in female adolescents with excess weight, with the latter possibly being the sole trigger. However, we were not able to corroborate this in our sample, as we did not find a significantly higher proportion of girls (55%) and only 25% of the patients had excess weight.
The aetiology of IIH is currently unknown, although there are several hypotheses on the subject. Different authors in the literature have reported a direct association with the use of different drugs or supplements that are potential triggers, such as corticosteroids, GH (a risk factor present in 5% of our sample), vitamin A (hypervitaminosis) or tetracyclines, and with dural sinus malformations (present in 1 patient in our sample).3,4
The most frequent presenting complaint is headache associated with vomiting or changes in vision (blurred vision due to papilloedema or double vision due to sixth cranial nerve palsy). In our sample, the key clinical feature was holocranial tension headache, with was associated with visual impairment in 22% of cases. A minority of patients presented with bilateral papilloedema that was a chance finding in a routine vision checkup, with no other symptoms.
Idiopathic intracranial hypertension is a diagnosis of exclusion that requires ruling out other possible causes of raised intracranial pressure, such as encephalitis or meningitis, venous thrombosis or, although much rarer, leptomeningeal carcinomatosis or neurocutaneous melanosis, among others.
The aim of treatment is to achieve symptom resolution and preserve visual function, preventing potential sequelae by decreasing intracranial pressure.
In our sample, in the 2 patients in whom IIH was associated with treatment with GH, the latter was suspended, which achieved resolution of symptoms. In all other patients, pharmacological treatment was initiated with acetazolamide as the first-line drug at 25-30 mg/kg/day divided in 2 doses. Acetazolamide is a carbonic anhydrase inhibitor that works by reducing the production of CSF, although this requires high doses that may be poorly tolerated, in which case it can be switched to furosemide or combined with a corticosteroid. Corticosteroids may be a useful temporizing measure in patients with rapid progression of visual impairment while surgical options are being considered. It is important that corticosteroids are discontinued after a gradual taper, as abrupt discontinuation can cause a rebound increase in intracranial pressure.
Other drugs are used in adult patients, such as topiramate and zonisamide, which are also carbonic anhydrase inhibitors. However, there are no data in the literature on their use in paediatrics.
The last tier of treatment involves surgery in cases with progressive impairment of visual function or headache refractory to pharmacological treatment. The invasive procedures that may be used in these patients include performance of repeated lumbar punctures or placement of lumboperitoneal or ventriculoperitoneal shunts, as well as interventions that target papilloedema, such as optic nerve sheath decompression or fenestration (although there is limited experience with this approach in children).
On reviewing the current medical literature, we did not find any reports of new approaches to treatment. The use of serial punctures for drainage has decreased significantly, as this is now considered unnecessarily invasive.5-7
Although the published data evinces a high rate of spontaneous resolution, we prefer to initiate pharmacological treatment early due to the risk of visual impairment.1
It is important that these patients undergo periodic clinical and ophthalmological evaluations. The latter should not be limited to fundoscopic examinations, but also visual field tests performed by an ophthalmologist, as abnormalities in the visual field are indicative of disease progression and worsening.
Once ophthalmologic findings become normal, pharmacological treatment should be tapered off to discontinuation. It is recommended that acetazolamide be tapered off over a period of at least 2 months. Patients should remain in followup after treatment discontinuation due to the risk of recurrence (8%-38% of patients): in our sample, we found recurrences in 15% of patients.
CONCLUSION
Idiopathic intracranial hypertension is an infrequent disease, but one whose early diagnosis and treatment is essential to prevent potentially irreversible sequelae. It should be managed by a well-coordinated multidisciplinary care team including paediatric neurologists, ophthalmologists and neurosurgeons.
We ought to emphasise the importance of the ophthalmological evaluation, including a fundoscopic examination and visual field test, as it is essential for diagnosis and followup and to determine how aggressive treatment needs to be.
It is also important that the unit has a protocol in place for diagnosis and treatment that can guide the performance of diagnostic tests and therapeutic decision-making in these patients.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare in relation to the preparation and publication of this article.
ABREVIATURAS
CSF: cerebrospinal fluid · CT: computed tomography · EEG: electroencephalogram · GH: growth hormone · IIH: idiopathic intracranial hypertension · MRI: magnetic resonance imaging.
REFERENCES
- Monge Galindo L, Pérez Delgado R, López-Pisón J, Olloqui-Escalona A, García Íñiguez JP, Ruiz del Olmo Izuzquiza L. Hipertensión intracraneal benigna. Experiencia en 18 años. An Pediatr (Barc). 2009;71:400-6.
- Mosquera Gorostidi A, Iridoy Zulet M, Azcona Ganuza G, Gembero Esarte E. Seudotumor cerebri en niños: etiología, características clínicas y evolución. Neurologia. 2017.pii: S0213-4853(16)30244-4.
- Eldes NH, Yilmaz Y. Pseudotumour cerebri in children: etiological, clinical features and treatment modalities. Eur J Paediatr Neurol. 2012;16:349-55.
- Ray WZ, Lee A, Blackburn SL, Lueder GT, Leonard J R. Pseudotumor cerebri following tapered corticosteroid treatment in an 8-month-old infant. J Neurosurg Pediatr. 2008;1:88-90.
- Rodríguez de Rivera FJ, Martínez-Sánchez P, Ojeda-Ruiz de Luna J, Arpa-Gutiérrez FJ, Barreiro-Tella P. Benign intracranial hypertension. History, clinical features and treatment in a series of 41 patients. Rev Neurol. 2003;37:801-5.
- Aylward SC, Way AL. Pediatric intracranial hypertension: a current literature review. Curr Pain Headache Rep. 2018;22:14.
- Youroukos S, Psychou F, Fryssiras S, Paikos P, Nicolaidou P. Idiopathic intracranial hypertension in children. J Child Neurol. 2000;15:453-7.
- Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-65.
- Lee EB, Edelman FS, Stafstrom CE. Evidence of diplopia in children’s headache drawings helps to differentiate pseudotumor cerebri from migraine. Pediatr Neurol. 2018;79:40-4.
- Rangel-Castillo L, Robertson C. Management of intracranial hypertension. Critical Care Clin. 2006;22:713-32.
Comments
This article has no comments yet.