
Vol. 15 - Num. 57
Notas clínicas
Casos clínicos en Endocrinología (n.º 1): niña de seis años con pubarquia
Noelia Álvarez Gila, Fernando Sánchez Peralesb
aEndocrinología infantil. Servicio de Pediatría. Hospital de Torrejón. Torrejón de Ardoz. Madrid. España.
bServicio de Pediatría. Hospital de Torrejón. Madrid. España.
Correspondencia: N Álvarez. Correo electrónico: noelia.alvarez.gil@gmail.com
Cómo citar este artículo: Álvarez Gil N, Sánchez Perales F. Casos clínicos en Endocrinología (n.º 1): niña de seis años con pubarquia. Rev Pediatr Aten Primaria. 2013;15:73-80.
Publicado en Internet: 01-04-2013 - Número de visitas: 44719
Resumen
Comenzamos con este artículo la publicación de una serie de casos en Endocrinología pediátrica que constituyen un motivo de consulta frecuente en Atención Primaria y que, aunque la mayoría de las veces tienen que ser derivados a Atención Especializada, es interesante conocer tanto el diagnóstico final como el tratamiento. En este primer caso, se trata de una niña de seis años con pubarquia. Se discute el diagnóstico diferencial y el tratamiento.
Palabras clave
● Hiperplasia suprarrenal congénita ● PubarquiaCASO CLÍNICO
Niña de seis años y nueve meses que acude a consulta remitida por su pediatra de Atención Primaria (AP), por pubarquia precoz detectada en la revisión de los seis años. No se ha objetivado axilarquia, telarquia ni olor apocrino. Los padres refieren un aumento de la velocidad de crecimiento, ya que, desde muy pequeña, se encuentra en percentiles elevados de talla y es más alta que su hermano mellizo.
Sigue una alimentación adecuada y es una niña activa y con buen rendimiento escolar. Como antecedentes personales, se trata de una niña nacida de embarazo gemelar bicorial biamniótico de padres no consanguíneos, a término, sin incidencias en el periodo neonatal. Cribado endocrinometabólico negativo. Sin otros antecedentes de interés.
No existen antecedentes familiares de desarrollo precoz, alteraciones de fertilidad, ni alteraciones menstruales; tampoco hirsutismo.
Somatometría
Peso: 28,2 kg (percentil 81 [P81]; +0,88 desviaciones estándar [DE]). Talla: 127 cm (P90; +1,3 DE). Índice de masa corporal (IMC): 17,48 kg/m2 (P64; +0,37 DE). Superficie corporal: 1 m2.
Evaluación de la talla adulta
Talla del padre: 180 cm (P66; +0,42 DE). Talla de la madre: 160 cm (P26; -0,66 DE). Talla diana: 163,5±5 cm (P47; -0,08 DE).
Exploración física
Fenotipo normal. Buen estado general. Bien hidratada y perfundida. Bien nutrida. Normocoloreada. Sin bocio ni tiroides palpable. Estadio puberal de Tanner: A1S1P2 (vello rizado oscuro en los labios mayores). Sin hipertrofia de clítoris. El resto de la exploración estaba dentro de la normalidad.
Pruebas complementarias
Se solicitan una radiografía de la muñeca izquierda, que evidencia una edad ósea de siete años y diez meses (+1,5 DE para la edad cronológica de seis años y nueve meses) y una analítica con hemograma, perfil férrico, hepático, tiroideo y renal. En la Tabla 1 se destacan los parámetros analíticos de mayor relevancia.
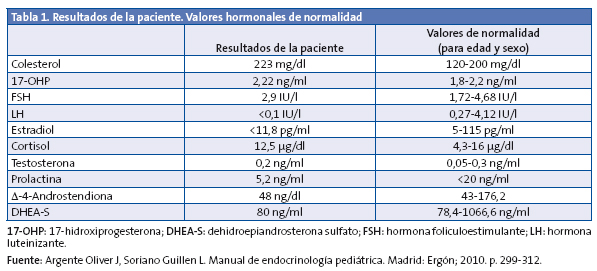
Ante la elevación de 17-hidroxiprogesterona (17-OHP), se realiza test de Synacthen®, administrando 250 µg de hormona adrenocorticotropa (ACTH) y midiendo el pico a los 60 minutos de 17-OHP, con resultado de 16,34 ng/dl y cortisol 32,38 µg/dl.
Cuando el resultado de 17-OHP es mayor de 10 ng/dl, es necesario realizar un estudio genético, obteniendo una muestra de ADN de sangre periférica. En este caso, se detecta una mutación en homocigosis (ambas cadenas de ADN) V281L situada en el exón 7, que supone una afección parcial de la actividad enzimática de 21-hidroxilasa (estudios in vitro la sitúan en un 25-75% de la actividad).
Diagnóstico inicial
Con estos resultados, la paciente es diagnosticada de hiperplasia suprarrenal, forma no clásica o tardía con mutación en homocigosis de V281L.
En el seguimiento, presenta una evolución de su pubarquia lentamente progresiva sin aparición de otros signos de pubertad precoz y sin aceleración importante de la edad ósea, con buena predicción de talla por el método Bayley-Pinneau: 165,2±5 cm, por lo que, por el momento, no se inicia tratamiento con glucocorticoides, realizándose solo seguimiento clínico.
COMENTARIOS
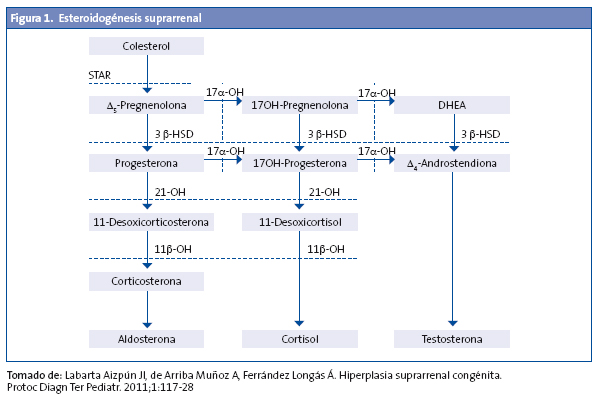
La hiperplasia suprarrenal congénita (HSC) comprende un grupo de enfermedades hereditarias autosómicas recesivas que afectan a la síntesis de esteroides suprarrenales, se caracteriza por un bloqueo total o parcial de la síntesis de cortisol y se acompaña en ocasiones, y en las formas graves, de un déficit de aldosterona.
El déficit de cortisol produce una retroalimentación negativa con elevación de la ACTH, que produce hiperestimulación de la corteza suprarrenal y aumento de los precursores previos al defecto enzimático1-3 (Fig. 1).
FORMAS CLÍNICAS
En función del grado de afectación enzimática, se dividen en formas graves y moderadas. En las formas graves o clásicas, el déficit es completo e inician sus manifestaciones en la época fetal-neonatal; en las formas moderadas o no clásicas, el déficit es parcial y se manifiestan clínicamente en la infancia y adolescencia, e incluso pueden pasar desapercibidas hasta la edad adulta.
Según la enzima afectada, se diferencian cinco formas clínicas:
- Déficit de 21-hidroxilasa.
- Déficit de 11ß-hidroxilasa.
- Déficit de 3ß-hidroxiesteroide-deshidrogenasa.
- Déficit de 17α-hidroxilasa (17a-OH).
- HSC lipoidea: déficit de la proteína StAR (steroidogenic acute regulatory Protein), proteína esencial para el transporte del colesterol al interior de la mitocondria y su posterior transformación en pregnenolona2,3.
El déficit de 21-hidroxilasa es el más frecuente, ya que corresponde a un 95% de los casos de HSC. Presenta dos características fundamentales: insuficiencia suprarrenal e hiperandrogenismo, que derivan directa o indirectamente, por un lado, en la incapacidad de transformar la 17-OHP en 11-desoxicortisol (déficit de secreción del cortisol) y la progesterona en desoxicorticosterona (déficit de secreción de aldosterona) y, por otro, en el acúmulo de 17-OHP, androstendiona, testosterona y sus metabolitos respectivos.
Existe una gran variabilidad clínica del déficit de 21-hidroxilasa ligada directamente al tipo de afectación de los alelos del gen.
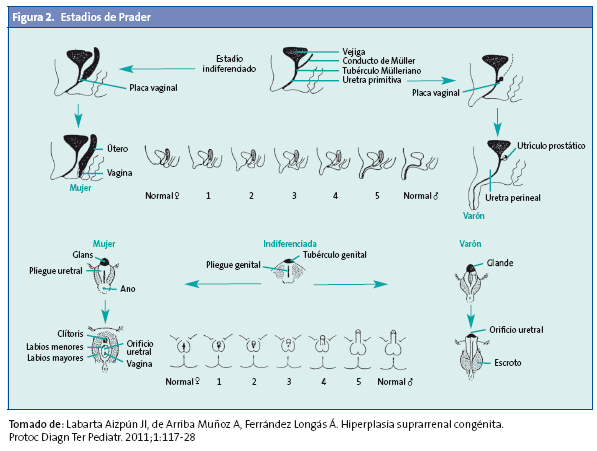
Las formas clásicas implican la existencia de un hiperandrogenismo que se puede expresar intraútero y que condiciona la aparición de macrogenitosomía en el varón y virilización variable de los genitales externos en la mujer (estadios de Prader) (Fig. 2) con manifestaciones desde una hipertrofia de clítoris aislada, hasta un grado máximo que puede determinar la asignación de sexo incorrecta como varón.
Dentro de las formas clásicas hay a su vez dos tipos:
- Forma con pérdida salina: es la expresión más grave de la enfermedad, con un déficit importante de cortisol y de aldosterona que se manifiesta en ambos sexos como crisis de pérdida salina aguda grave en la época neonatal que tiene una importante morbimortalidad si no se instaura un tratamiento adecuado.
- En la forma clásica virilizante simple, la afectación enzimática no es tan grave y hay un mínimo de actividad enzimática residual que determina que la síntesis de aldosterona y de cortisol no esté totalmente suprimida, por lo que no presentan crisis de pérdida salina. Las niñas son identificadas precozmente por la virilización de los genitales externos, pero los niños y aquellas niñas con una virilización leve suelen diagnosticarse tardíamente en la infancia cuando se hacen manifiestos los signos de hiperandrogenismo y la aparición de una pseudopubertad precoz.
En las formas no clásicas existe un hiperandrogenismo de aparición posnatal y pueden ser sintomáticas, o no sintomáticas o crípticas:
- En las sintomáticas, los síntomas más frecuentes en la infancia son pubarquia prematura, piel grasa, acné, aceleración del crecimiento y de la edad ósea con afectación variable de la talla adulta, y en las niñas puede existir una moderada hipertrofia del clítoris. En la adolescencia y la edad adulta las mujeres pueden presentar irregularidades menstruales, hirsutismo, calvicie, ovario poliquístico, acné e infertilidad. Los varones afectos pueden presentar acné, oligospermia e infertilidad, pero la mayoría de las veces son asintomáticos.
- Las formas crípticas o no sintomáticas cursan únicamente con hallazgos hormonales, aunque pueden presentar eventualmente algún signo clínico de hiperandrogenismo1-5.
DIAGNÓSTICO
El diagnóstico hormonal se basa en la demostración de niveles plasmáticos elevados de 17-OHP. En el déficit clásico, la 17-OHP basal está muy elevada, mientras que en las formas no clásicas el bloqueo es menos grave y la acumulación de 17-OHP puede ser muy variable, siendo aconsejable la realización de un test de ACTH.
El diagnóstico genético consiste en determinar la anomalía en el gen CYP21A2 que codifica la enzima 21-hidroxilasa.
Se debería hacer un estudio familiar que permita el diagnóstico de portadores o de formas no clásicas oligosintomáticas y/o crípticas.
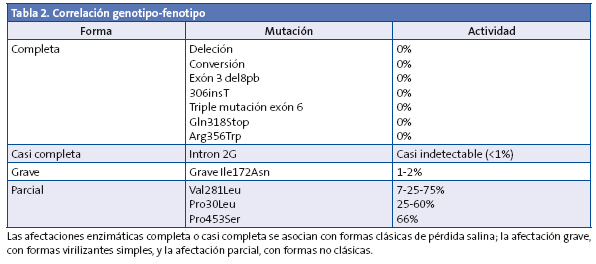
La correlación genotipo/fenotipo en el déficit de 21-hidroxilasa es muy intensa, ya que la gravedad de los signos clínicos deriva directamente del grado de déficit enzimático (Tabla 2)1,6,7.
Cribado neonatal: la hiperplasia suprarrenal congénita es una de las enfermedades metabólicas incluidas en el cribado del recién nacido en la mayoría de las comunidades autónomas.
TRATAMIENTO
Tratamiento sustitutivo con glucocorticoides
- Formas clásicas de déficit de 21-hidroxilasa deben ser tratadas con glucocorticoides (GC) para suprimir el exceso de secreción de hormona estimulante de la corticotrofina y ACTH, y reducir el exceso de esteroides sexuales de origen suprarrenal. La hidrocortisona es el tratamiento más fisiológico, al tener una potencia superponible a la del cortisol endógeno, si bien un tratamiento excesivo y precoz con GC es potencialmente nocivo para el crecimiento1,5,7,9,10.
- En los pacientes con HSC no clásica, el tratamiento se reservaría para las formas muy sintomáticas:
- Pubarquia rápidamente evolutiva con aceleración de edad ósea y pronóstico de talla final baja.
- Adolescentes mujeres con hirsutismo y acné grave.
- En adultos con hipofertilidad7.
Tratamiento sustitutivo con mineralocorticoides
Los pacientes con pérdida salina requieren la administración de un mineralocorticoide. El más utilizado es la 9-α-fluorhidrocortisona o fludrocortisona (Astonin®) y suplementos de cloruro de sodio (1-2 g por día) durante el primer año de vida.
También en la forma virilizante simple, aunque secretan una cantidad adecuada de aldosterona y no tienen crisis de pérdida salina, se necesita tratamiento para el control de las cifras elevadas de renina.
Una dosificación excesiva de 9α-fluorhidrocortisona también puede retrasar el crecimiento y producir hipertensión o taquicardia1.
Crisis de insuficiencia suprarrenal
Ante situaciones de estrés o enfermedades intercurrentes, los pacientes tienen riesgo de desarrollar insuficiencia suprarrenal aguda, por lo que es recomendable que lleven placa identificativa. En esas situaciones se debe duplicar o triplicar la dosis de mantenimiento de hidrocortisona oral y en caso de que se establezca una crisis suprarrenal es necesario un control hospitalario.
Tratamiento quirúrgico de los genitales ambiguos y apoyo psicológico
La actitud terapéutica se inicia con la asignación precoz del sexo, que debe ser la del sexo genético/gonadal, por la posibilidad de mantener la función reproductora. En la actualidad, se están obteniendo buenos resultados con la realización de la reconstrucción genital (clitoroplastia y vaginoplastia) en un mismo acto quirúrgico (hacia el segundo semestre de edad) siendo el objetivo la corrección completa de los genitales externos antes de los 18 meses de edad. Se debe ofrecer apoyo psicológico a las pacientes con ambigüedad genital y una información completa e individualizada a la familia1,5.
SEGUIMIENTO Y PAPEL DEL PEDIATRA DE ATENCIÓN PRIMARIA
El papel del pediatra de AP es fundamental en la detección de los signos iniciales, como la pubarquia precoz en este caso, pero también otros que hemos mencionado como piel grasa, acné o aceleración del crecimiento.
Para el control del tratamiento se utiliza la determinación de ?4 androstendiona, que presenta menos variación circadiana que la 17-OHP y debe hacerse en el nivel especializado.
Se necesita un control clínico adecuado de forma conjunta entre el pediatra endocrinólogo y el de AP, vigilando la velocidad de crecimiento, la maduración ósea, la curva de peso y el IMC1,3.
A largo plazo, ya en la edad adulta, habría que estar pendiente de la aparición de osteoporosis, infertilidad, hirsutismo, alteraciones menstruales en mujeres y la aparición de masas testiculares secundarias a tejido suprarrenal ectópico en los varones.
Por último, pero no menos importante, el pediatra de AP puede constituir un gran apoyo para el niño y la familia, reforzando la importancia del tratamiento y conociendo el pronóstico final, ya que las formas clásicas con adecuado tratamiento y control hormonal suelen tener una talla adulta alrededor de -1 DE para su talla genética y las formas no clásicas o tardías una talla final acorde con talla genética7,13.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS: 17-OHP: 17-hidroxiprogesterona • ACTH: hormona adrenocorticotropa • AP: Atención Primaria • DE: desviación estándar • GC: glucocorticoides • HSC: hiperplasia suprarrenal congénita • IMC: índice de masa corporal • Pn: percentil n.
BIBLIOGRAFÍA
- Labarta Aizpún JI, de Arriba Muñoz A, Ferrández Longás Á. Hiperplasia suprarrenal congénita. Protoc Diagn Ter Pediatr. 2011;1:117-28. Disponible en http://www.aeped.es/sites/default/files/documentos/10_hiperplasia_suprarrenal_congenita.pdf (consultado el 21/01/2013).
- Oliver A, Ezquieta B, Gussinyé M. Hiperplasia suprarrenal congénita. En: Argente J, Carrascosa A, Gracia R, Rodríguez Hierro F, eds. Tratado de Endocrinología Pediátrica y de la Adolescencia. 2.ª edición. Barcelona: Doyma; 2000. p. 995-1042.
- Sánchez Bachena T. Hiperplasia suprarrenal congénita. En: Pombo. Tratado de endocrinología pediátrica. 4.ª ed. Ciudad: Editorial; 2009.
- Antal Z, Zhou P. Congenital adrenal hyperplasia: diagnosis, evaluation, and management. Pediatr Rev. 2009;30:49-57.
- Speiser W, Azizz R, Baskin L, Ghyzzoni L, Hensel T. Congenital adrenal hyperplasia due to steroid 21 hydroxilase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(9):4133-60.
- Ezquieta Zubicaray B, Cueva López E, Varela Junquera JM, Jariego Fente C. Aportaciones del análisis molecular en la hiperplasia suprarrenal congénita. Acta Pediatr Esp. 2001;59:479-96.
- Alonso M, Ezquieta B. Hiperplasia suprarenal congénita no clásica o tardía. Rev Esp Endocrinol Pediatr. 2012;3(Suppl).
- Labarta JI, Bello E, Ruiz-Echarri, Rueda C, Martul P. Estado en la edad adulta y propuesta de optimización terapéutica de la hiperplasia suprarrenal congénita. An Pediatr. 2003;58(Supl 2):12-34.
- Hindmarsh PC. Management of the child with congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:193-208.
- Nieman Lk. Diagnosis and treatment of nonclassic (late-onset) congenital adrenal hyperplasia due to 21-hidroxylase. Up to Date, 2010.
- Quintos JB, Vogiatzi MG, Harbison MD, New MI. Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001:86:1511-7.
- Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improve final adult height in children congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2005;90(6):3318-25.
- Muthusamy K, Elamin M, Smishkin G. Adult height in patients with congenital adrenal hyperplasia: a systemic review and metanalisis. J Clin Endocrinol Metab. 2010;151:U77-U82.