

Vol. 15 - Num. 60
Notas clínicas
Recién nacida con flictenas
Paola Gómez Rodrígueza, C López Calerob
aMIR-MFyC. CS Mairena del Aljarafe. Mairena del Aljarafe. Sevilla. España.
bPediatra. CS Mairena del Aljarafe. Mairena del Aljarafe. Sevilla. España.
Correspondencia: P Gómez. Correo electrónico: paola19_677@hotmail.com
Cómo citar este artículo: Gómez Rodríguez P, López Calero C. Recién nacida con flictenas. Rev Pediatr Aten Primaria. 2013;15:333-6.
Publicado en Internet: 13-12-2013 - Número de visitas: 16513
Resumen
Presentamos el caso de una recién nacida con flictenas a las 48 horas de vida, que al cicatrizar presentan punteado blanquecino en forma de quistes de milium, por lo que se sospecha la posibilidad de una enfermedad ampollosa congénita.
Finalmente, el Servicio de Dermatología confirma el diagnostico de epidermólisis ampollosa de Weber-Cockayne.
Palabras clave
● Ampollas ● Congénito ● Epidermólisis ampollosa ● Pequeños traumatismos ● Quistes de milium ● Recién nacidoCASO CLÍNICO
Recién nacida que, a las 48 horas de vida, presenta flictenas en el dorso del pie (Fig. 1), donde previamente tenía la pulsera de identificación del recién nacido; aunque también las presenta en el pie contralateral y algunas pequeñas en el dorso de una de las manos (Fig. 2).
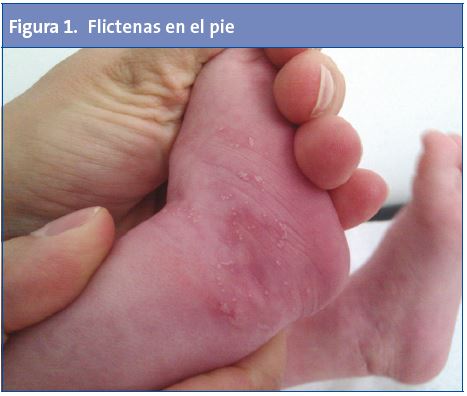
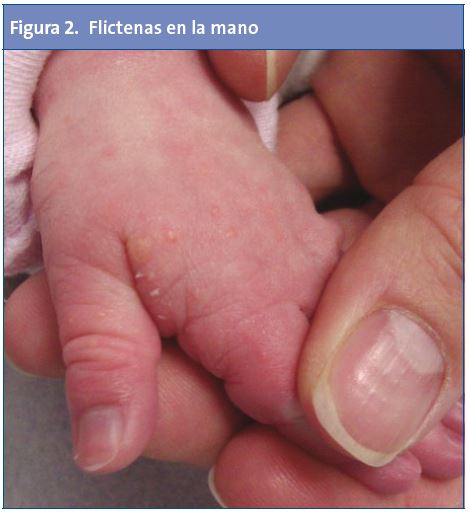
Como antecedentes personales, tan solo presenta frenillo sublingual y oblicuidad pélvica congénita. Tanto el embarazo como el parto fueron normales. En sus antecedentes familiares no figuran enfermedades ampollosas conocidas ni otros datos de interés, salvo un tío materno con hemofilia.
En la exploración física, además de las flictenas ya mencionadas, presenta buen estado general, con palidez de piel y mucosas. Fototipo I. Limitación a la abducción de la cadera izquierda, asimetría en huecos poplíteos y signo de Galeazzi positivo. Frenillo sublingual. El resto de la exploración está dentro de la normalidad.
Se trata como un impétigo ampolloso con fomentos de sulfato de zinc al 1/1000 tres veces al día y amoxicilina-clavulánico 100-12,5 suspensión oral en dosis de amoxicilina de 50 mg/kg/día repartidas en tres dosis durante siete días.
Al cicatrizar las lesiones, se objetiva un punteado blanquecino en forma de quistes de milium en la zona de la flictena, por lo que se sospecha la posibilidad de una enfermedad ampollosa congénita.
A los cuatro meses, reaparecen de nuevo las lesiones ampollosas en los pies y en la pierna izquierda al mínimo roce, asociándose a sudoración en palmas y plantas.
Se derivó a la paciente al Servicio de Dermatología, donde se confirmó la sospecha de enfermedad ampollosa congénita o epidermólisis ampollosa de Weber-Cockayne.
Se realiza una búsqueda bibliográfica con tres palabras clave: epidermólisis ampollosa, recién nacido y cicatrización en quistes de milium.
COMENTARIOS
La epidermólisis bullosa o ampollosa hereditaria es una genodermatosis de baja prevalencia, transmitida de forma autosómica dominante o recesiva y causada por una alteración de las proteínas de la unión epidermodérmica, lo que da lugar a la formación de ampollas y erosiones cutáneas y mucosas al mínimo trauma o roce. Se clasifica en tres grupos:
- Epidermólisis ampollosa simple o bullosa (EAS):causada por mutaciones en los genes que codifican la queratina 5 o 14, y producen ampollas intraepidérmicas que curan sin cicatriz. Se dividen en:
- EAS de Weber-Cockayne: las lesiones iniciales aparecen durante la infancia o adolescencia. Ocurren tras traumatismos y en la época estival con mayor frecuencia. Las lesiones se localizan, típicamente, en palmas y plantas, lo cual, unido a una frecuente hiperhidrosis, a veces produce disfuncionalidad manual o para la marcha. La cicatrización de las lesiones es completa, sin mostrar atrofia, quistes de milium o pigmentaciones.
- EAS de Koebner: es más precoz, con ampollas generalizadas y formación de quistes de milium.
- EAS de Dowling-Meara: con distribución herpetiforme y posible aparición aguda.
- EAS con distrofia muscular y ampollas localizadas.
- Epidermólisis ampollosa de la unión (EAU): las ampollas se deben a una formación anormal de los componentes de los hemidesmosomas, con roturas por la lámina lúcida. Se dividen en:
- EAU de Herlitz.
- EAU no Herlitz.
- EAU con atresia pilórica.
- Epidermólisis ampollosa distrófica (EAD):la alteración del colágeno VII impide el anclaje de la epidermis, produciéndose ampollas subepidérmicas que se reparan con cicatrices. Se dividen en:
- EAD dominante, con los tipos de Cockayne-Touraine y de Pacini.
- EAU recesiva, de tipo Hallopeau-Siemens.
Clínica
La clínica fundamental es la presencia de ampollas que aparecen al mínimo traumatismo o roce. El cuadro comienza desde el nacimiento o más adelante en la vida, siendo en general de peor pronóstico cuanto más precoces. Las lesiones se pueden reparar sin ninguna secuela o dejar cicatrices, causar quistes de milium y motivar adherencias y deformidades.
La sobreinfección bacteriana es la principal complicación de todas las formas de epidermólisis bullosa.
La realización de una exploración cuidadosa, atendiendo al aspecto de las lesiones, olor, dolor, fiebre, permitirá la sospecha de infección.
Diagnóstico
El diagnóstico se realiza basándose en los aspectos clínicos, histopatológicos, biopatológicos y genéticos.
Desde el punto de vista clínico, la exploración del niño afectado por un cuadro ampolloso debe ser completa y lo más exhaustiva posible. Es muy importante la historia clínica detallada, haciendo especial hincapié en los antecedentes familiares.
Histopatológicamente, es imprescindible la realización de una biopsia cutánea para examen al microscopio óptico, con tinción de hematoxilina y eosina e inmunohistoquímica, y al microscopio electrónico. La biopsia cutánea debe ser realizada sobre piel sana, previamente frotada, de cualquier parte del tegumento paralesional en las generalizadas o perilesional en el caso de las localizadas. Se considera adecuada la obtención de tres cilindros cutáneos mediante la técnica de biopsias-punch.
Diagnóstico diferencial
El diagnóstico diferencial debe hacerse con todos los procesos cutáneos cuya lesión elemental sea la ampolla. Debemos pensar también en dermatitis herpetiforme infantil, enfermedad ampollosa por IgA lineal, e impétigo ampolloso.
Histopatológicamente, el diagnóstico diferencial se realiza con la epidermólisis ampollosa adquirida clásica y la porfiria cutánea tarda.
Tratamiento
No existe aún tratamiento definitivo. El tratamiento de estos procesos es paliativo; quienes los padecen deben usar ropa y calzado adecuados, evitar actividades con alto riesgo de motivar ampollas y realizar cuidados adecuados de las mismas cuando se vayan produciendo.
En la epidermólisis bullosa simple debe tratarse la hiperhidrosis que acompaña a muchas de estas formas, pues es la principal causa de aparición de las ampollas. Se aplican sales de aluminio vía tópica (cloruro de aluminio al 20-35%).
Las uñas deben cuidarse con el uso de jabones antisépticos y pomadas antibióticas, para evitar la sobreinfección.
Debemos tener en cuenta también el consejo genético y las técnicas de diagnóstico prenatal, que deben ser tratados con los padres, y siempre dentro de las normas de la Bioética y los aspectos médico-legales.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS:EAD: epidermólisis ampollosa distrófica • EAS: epidermólisis ampollosa simple • EAU: epidermólisis ampollosa de la unión.
BIBLIOGRAFÍA
- Lloyd C, Yu QC, Cheng J, Turksen K, Degenstein L, Hutton E, et al. The basal keratin network of stratified squamous epithelia: defining K15 function in the absence of K14. J Cell Biol. 1995;129:1329-44.
- Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A, et al. Revised classification system for inherited epidermolysis bullosa: report of the second international consensus meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42:1051-66.
- Uitto J, Eady R, Fine JD, Feder M, Dart J. The DEBRA International Visioning/Consensus Meeting on Epidermolysis Bullosa: summary and recommendations. J Invest Dermatol. 2000;114:734-7.
- Bermejo E, Marco J, Paisán L. Epidermólisis Bullosa: Patogénesis, aspectos clínicos, diagnósticos y genéticos, base molecular, aspectos epidemiológicos, manejo del paciente con EB e implicaciones translacionales del análisis de mutaciones. Rev Dismorfología y Epidemiología. 2005;V(4).
- Izquierdo M, Avellaneda A. Enfermedades raras; un enfoque práctico. Madrid: Instituto de Investigación de Enfermedades Raras, Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo; 2004 [en línea] [consultado el 19/01/2013]. Disponible en http://goo.gl/75kZK
- Tidman MJ, Garzon MC. Vesiculobullous diseases. En: Schachner LA, Hansen RL. Pediatric Dermatology. London: Mosby; 2003. p. 683-712.
- Fe JD. Epidermólisis ampollosa. En: Bolognia JL, Jorizzo JL, Rapini RP. Dermatología (vol. 1). Madrid: Elsevier España; 2004. p. 491-500.
Comentarios
Este artículo aún no tiene comentarios.