
Vol. 23 - Num. 90
Colaboraciones especiales
Cribado neonatal de la atrofia muscular espinal: el momento es ahora
Lucía López Granadosa, François Boemerb, Tatiana Pereirac, Laurent Servaisd, Ingrid Moralese
aPediatra. Office de la Naissance et de l’Enfance (ONE). Bruselas. Bélgica.
bLaboratorio de Bioquímica Genética. Departamento de Genética Humana. CHU Lieja. Universidad de Lieja. Bélgica.
cEnfermera. Gestión de programas de salud. Office de la Naissance et de l’Enfance. Bruselas. Bélgica.
dNeuropediatría. División de Neurología pediátrica. Centro de referencia de Enfermedades Neuromusculares. Departamento de Pediatría. CHU, Lieja, Bélgica. MDUK Neuromuscular Centre, Departamento de Pediatría, Universidad de Oxford. Reino Unido.
eOffice de la Naissance et de l’Enfance (ONE). Bruselas. Bélgica .
Correspondencia: L López. Correo electrónico: luciagranados@yahoo.es
Cómo citar este artículo: López Granados L, Boemer F, Pereira T, Servais L, Morales I. Cribado neonatal de la atrofia muscular espinal: el momento es ahora. Rev Pediatr Aten Primaria. 2021;23:211-4.
Publicado en Internet: 21-06-2021 - Número de visitas: 11949
Resumen
La atrofia muscular espinal (AME) es la enfermedad neurodegenerativa más común en la infancia. La aprobación reciente de nuevas terapias efectivas ha justificado la implementación desde hace dos años de un programa piloto de cribado neonatal en la Federación Valonia Bruselas (FWB).
Se describe el procedimiento de cribado neonatal utilizado actualmente para detección para la AME en recién nacidos en el sur de Bélgica. El coste del cribado es de 3,10 € por niño. Si se mantiene esta configuración, representa un coste anual de 181 000 € (por aproximadamente 60 000 nacimientos anuales en FWB).
En el contexto de contar con un método de cribado neonatal fiable, con un coste económico moderado y la posibilidad de un tratamiento inmediato viable, los autores recomiendan la inclusión y financiación del despistaje de la atrofia muscular espinal en la lista de enfermedades incluidas oficialmente en el marco del cribado neonatal.
Palabras clave
● Atrofia muscular espinal ● Cribado neonatal ● Enfermedad de Werdnig-Hoffman ● SMN1INTRODUCCIÓN
La atrofia muscular espinal (AME) es una enfermedad autosómica recesiva causada por mutaciones en el gen de la neurona motora de supervivencia (SMN1). Es la enfermedad neurodegenerativa más común en la infancia.
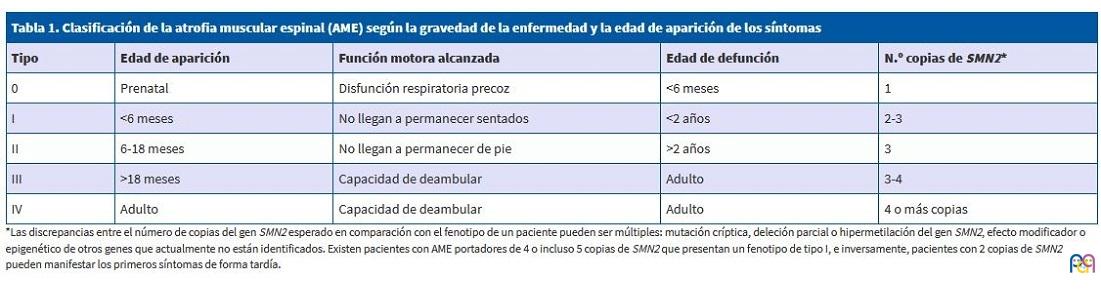
La enfermedad se asocia, en el 95% de los casos, a una deleción homocigótica del exón 7 del gen SMN1. La gravedad clínica está estrechamente relacionada con la presencia de un segundo gen, SMN2, quién produce aproximadamente un 10% de la proteína SMN funcional. El fenotipo será en general más benigno cuantas más copias de SMN2 haya.
La enfermedad, sin tratamiento, se presenta como una pérdida de fuerza muscular con aparición de una parálisis progresiva. Los fenotipos clínicos se agrupan en cinco formas diferentes, según la gravedad de la enfermedad y la edad de aparición (Tabla 1).
| Tabla 1. Clasificación de la atrofia muscular espinal (AME) según la gravedad de la enfermedad y la edad de aparición de los síntomas | ||||
|---|---|---|---|---|
| Tipo | Edad de aparición | Función motora alcanzada | Edad de defunción | N.º copias de SMN2* |
| 0 | Prenatal | Disfunción respiratoria precoz | <6 meses | 1 |
| I | <6 meses | No llegan a permanecer sentados | <2 años | 2-3 |
| II | 6-18 meses | No llegan a permanecer de pie | >2 años | 3 |
| III | >18 meses | Capacidad de deambular | Adulto | 3-4 |
| IV | Adulto | Capacidad de deambular | Adulto | 4 o más copias |
Recientemente se han aprobado fármacos innovadores, dirigidos a SMN2 o destinados a reemplazar SMN1. La primera opción terapéutica es Nusinersen (aprobado en Europa en 2017). Es un fármaco antisentido (molécula complementaria a una cadena de ARN específica y que permite modular cuantitativa o cualitativamente su lectura) inyectable a nivel intratecal que ha demostrado ser eficaz en los tipos 11, y también en los tipos 2 y 32. Los estudios han demostrado un aumento en la supervivencia y en la adquisición de nuevas habilidades motoras. Los indicadores mejoran aún más cuando el tratamiento se administra de forma temprana e idealmente antes de que aparezcan los síntomas3. Un segundo fármaco, Risdiplam, que permite una mejor expresión de la proteína SMN del gen SMN2, ha sido aprobado en EE. UU. en agosto de 2020.
Paralelamente, en 2019-2020, los ensayos de terapia génica han demostrado su eficacia en recién nacidos en fase presintomática de la enfermedad. Se trata de una inyección intravenosa de una única dosis de un virus atenuado en el que se ha insertado un gen funcional (Onasemnogene abeparvovec)4. Este tratamiento ha sido aprobado por la Food and Drug Administration y por la Agencia Europea del Medicamento en junio de 2020.
En el contexto de una enfermedad grave que causa una discapacidad grave en pacientes tratados tardíamente y que puede ser fatal sin tratamiento, el cribado en los recién nacidos parece la mejor solución para optimizar el efecto de las terapias innovadoras que modifican el pronóstico de los pacientes con AME.
La atrofia muscular espinal es cribada en la Federacion Valonia Bruselas (FWB) desde marzo de 2018 como parte del proyecto piloto Sun May Arise on SMA5. Para el establecimiento del programa se creó un comité encargado de coordinar las diversas acciones a realizar. Fueron precisos múltiples contactos con las autoridades políticas de la FWB, así como con las autoridades médicas (Consejo Superior de Genética, Office de la Naissance et de l’Enfance) y éticos, para garantizar su compromiso con este proyecto. Se implementó además un canal de comunicación al público vía Facebook (www.facebook.com/sunmayariseonsma/).
Varias fuentes de financiación, tanto del ámbito privado como público y asociativo contribuyeron al rápido despliegue del programa.
La metodología analítica se basa en una prueba qPCR del gen SMN1 en el ADN extraído en la prueba del talón del recién nacido6, sin que sea necesaria una recolección de sangre adicional. El genotipado del gen SMN1 se diseñó para detectar únicamente deleciones homocigóticas del exón 7 con una sonda de ácido nucleico específica. El método no identifica portadores heterocigotos de la deleción, mutaciones puntuales de SMN1 o el número de copias del gen modificador de SMN2. El desarrollo del método analítico se basó en informes previos de proyectos piloto realizados EE. UU. y Taiwán.
Las tarjetas de papel secante recogidas entre las 48 y 96 horas de vida se envían a los centros de despistaje seleccionados donde se efectúan los análisis (Fig. 1).
| Figura 1. Algoritmo de cribado neonatal para la atrofia muscular espinal en la Federación Valonia-Bruselas |
|---|
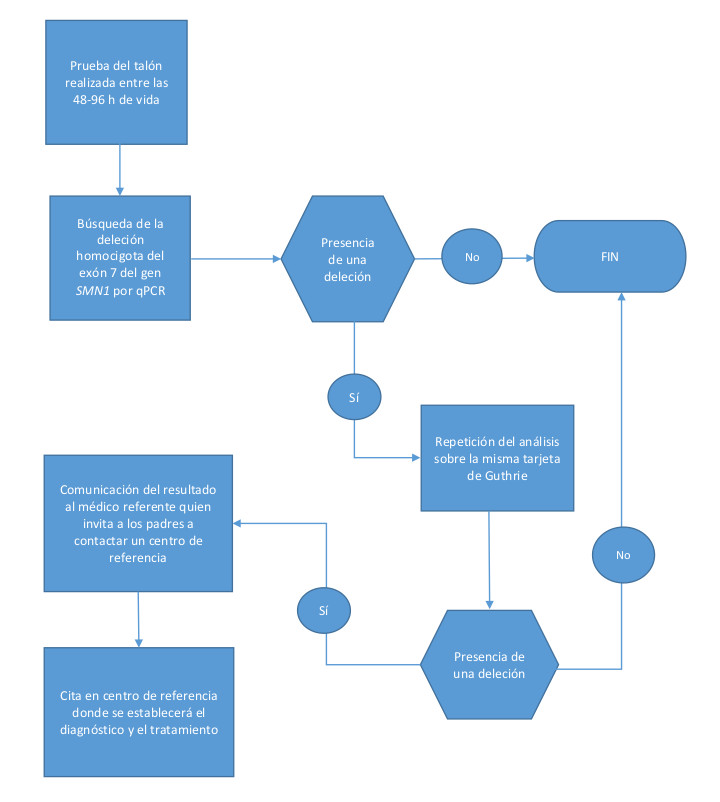 |
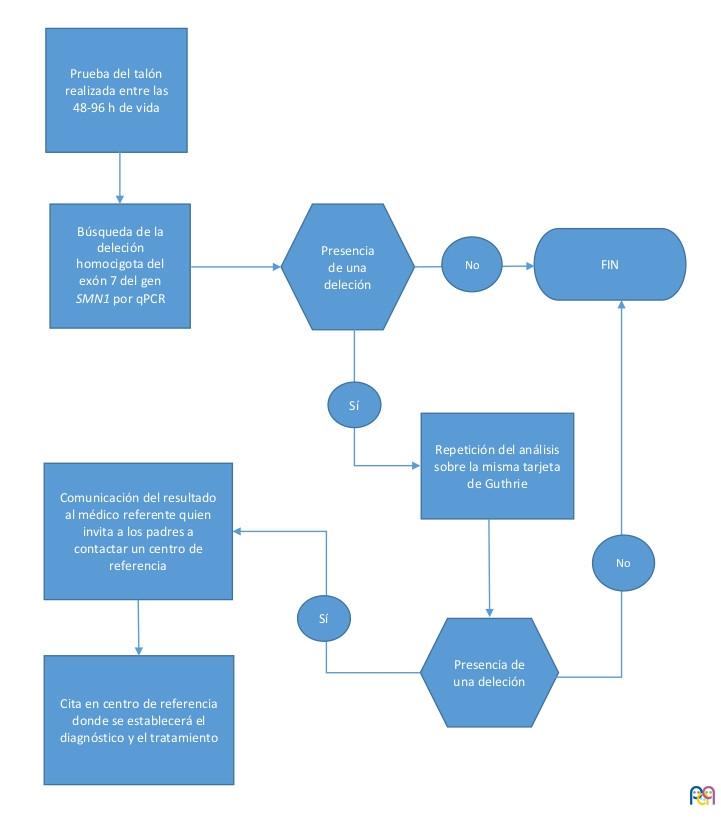
En el caso de un resultado positivo para la AME se informa simultáneamente al pediatra y al neurólogo referente. Los padres son contactados el mismo día por el neuropediatra de referencia, y se planifica una consulta lo antes posible para iniciar las pruebas confirmatorias utilizando una técnica alternativa. La decisión de los padres de incluir a su hijo en cualquier protocolo de tratamiento se basará en la información proporcionada por el neuropediatra.
El coste del cribado es de 3,10 € por niño. Si se mantiene esta configuración, representa un coste anual de 181 000 € (por aproximadamente 60 000 nacimientos anuales en FWB).
En el contexto de disponer de un método de despistaje neonatal fiable, con un coste económico moderado y la posibilidad de un tratamiento inmediato viable, los autores recomiendan la inclusión y el financiamiento de la detección la atrofia muscular espinal en la lista de enfermedades detectables en el marco del programa oficial de cribado neonatal.
CONFLICTO DE INTERESES
Laurent Servais es miembro de los consejos asesores de Biogen, AveXis, Roche y Cytokinetics Scientific y ha asesorado a Roche, Avexis y Biogen. Los demás autores no presentan conflictos de intereses.
ABREVIATURAS
AME: atrofia muscular espinal · FWB: Federación Valonia Bruselas.
FINANCIACIÓN
El estudio piloto de cribado neonatal en la Federación Valonia-Bruselas cuenta con el apoyo de Biogen, AveXis y la Association Belge contre les Maladies neuroMusculaires y del Ministerio de la Cultura, Infancia y Educación de la Comunidad Valonia-Bruselas.
BIBLIOGRAFÍA
- Aragon-Gawinska K, Seferian AM, Daron A, Gargaun E, Vuillerot C, Cances C, et al. Nusinersen in patients older than 7 months with spinal muscular atrophy type 1: A cohort study. Neurology. 2018;91:e1312-8.
- Mercuri E, Darras BT, Chiriboga CA, Chiriboga A, Day JW, Campbell C, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378:625-35.
- Dangouloff T, Servais L. Clinical Evidence Supporting Early Treatment Of Patients With Spinal Muscular Atrophy: Current Perspectives. Ther Clin Risk Manag. 2019;15:1153-61.
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017;377:1713-22.
- Boemer F, Caberg JH, Dideberg V, Beckers P, Marie S, Marcelis L, et al. (S)un (M)ay (A)rise on SMA: the hope of a region without spinal muscular atrophy. Rev Méd Liege. 2019;74:461-4.
- Boemer F, Caberg JH, Dideberg V, Dardenne D, Bours V, Hiligsmann M, et al. Newborn screening for SMA in Southern Belgium. Neuromuscul Disord. 2019;29:343-9.