

Vol. 27 - Num. 105
Notas clínicas
A raíz de un soplo inocente…
aMédico de Familia. CS Los Yébenes. Madrid. España.
Correspondencia: P Martínez. Correo electrónico: patriciamarias@hotmail.com
Cómo citar este artículo: Martínez Arias P. A raíz de un soplo inocente… . Rev Pediatr Aten Primaria. 2025;27:71-5. https://doi.org/10.60147/8d060abe
Publicado en Internet: 10-02-2025 - Número de visitas: 5387
Resumen
La miocardiopatía hipertrófica en la infancia es el segundo tipo de miocardiopatía, después de la miocardiopatía dilatada. Tiene mejor pronóstico, pero presenta riesgo de muerte súbita. Las causas más frecuentes son genéticas y hay síndromes específicos que cursan con MCH. Suele implicar alteraciones cardiacas mecánicas y riesgo de arritmias. Sin embargo, es frecuente que el niño esté asintomático y se le estudie por otro motivo, por ejemplo, a raíz de un cribado familiar o por alguna alteración detectada mediante pruebas complementarias. Se realiza el diagnóstico por imagen y el tratamiento puede implicar el implante de un desfibrilador.
Palabras clave
● Arritmia ● Miocardiopatía hipertrófica ● Soplo inocenteINTRODUCCIÓN
La miocardiopatía hipertrófica (MCH) no es un diagnóstico fácil inicialmente. El paciente puede estar asintomático y no sospecharse hasta fases avanzadas de la enfermedad, o debutar mediante muerte súbita. En esta ocasión, se revisa un caso pediátrico de un adolescente que acude a consulta de revisión del programa del niño sano y a raíz de un soplo inocente se llega al diagnóstico de MCH, abriendo el debate sobre el planteamiento del ECG de cribado en niños aparentemente sanos.
CASO CLÍNICO
Se trata de un adolescente que acude con sus padres a la revisión de los 14 años.
En la exploración física presenta buen estado general, está bien hidratado y perfundido, tiene pulsos periféricos simétricos y palpables, y en la auscultación cardiaca está rítmico, apreciándose un soplo en borde esternal izquierdo grado II/VI. La auscultación pulmonar no tiene ruidos sobreañadidos. El abdomen es normal, sin masas ni megalias. La tensión arterial arroja unas cifras de 105/60 mmHg.
Ante dicho soplo, de características aparentemente funcionales, se realiza un electrocardiograma en el centro de salud (Figura 1), en el que destaca una taquicardia sinusal a 150 lpm, desviación del eje a -30°, QTc de 0,49 segundos y signos de hipertrofia ventricular izquierda (HVI).
| Figura 1. Electrocardiograma realizado en el centro de salud con signos de hipertrofia ventricular izquierda |
|---|
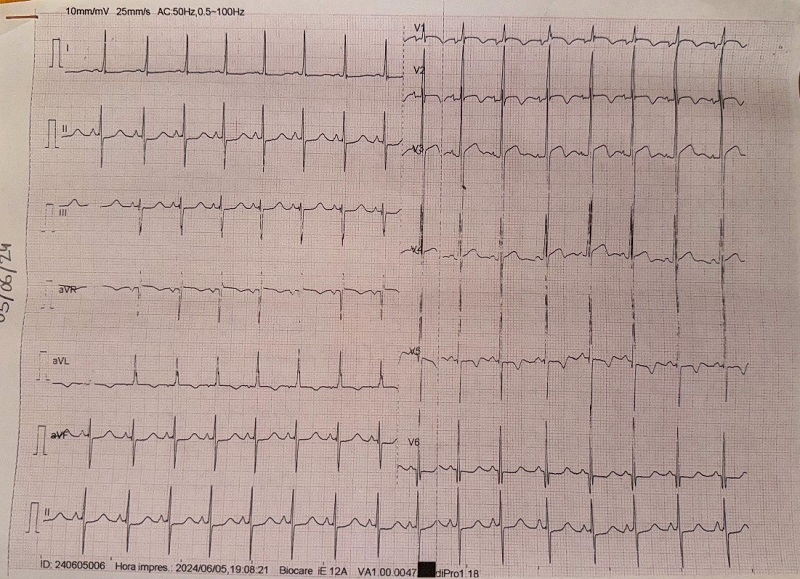 |

Ante estos hallazgos se indaga sobre síntomas cardiológicos, pero ni el niño ni los padres recuerdan episodios de dolor torácico, síncopes o palpitaciones. Realiza actividad física sin limitaciones (educación física en colegio y hockey 2 horas a la semana).
Se realiza derivación preferente a Cardiología, donde se efectúan las siguientes pruebas:
- Ecocardiograma: evidencia ventrículo derecho (VD) hipertrófico con función conservada y cavidad de VI disminuida por hipertrofia ventricular concéntrica severa con función sistólica conservada y alteración de función diastólica. No hay obstrucción del tracto de salida.
- Holter ECG: ritmo sinusal con frecuencia cardiaca media de 78 lpm, mínima de 40 y máxima de 140 lpm. Impresiona de QT largo, alteraciones de la repolarización con depresión significativa del ST, arritmia sinusal, y ausencia de ectopias. Está asintomático durante el registro.
- Ergometría: sin arritmias. No hay ascensos ni descensos patológicos del ST. Buena capacidad funcional (17,3 METS).
- Analítica normal, salvo un proBNP de 2695,74 ng/l (normal <125).
- Resonancia magnética (RM) (Figura 2): hipertrofia concéntrica severa del ventrículo izquierdo (VI) con septo de 31 mm. Función sistólica global en límites bajos de la normalidad. Presenta realce tardío puntiforme en septo basal y medio intramiocárdico junto con realce difuso global que afecta a la práctica totalidad del subendocardio del VI, siendo prácticamente transmural a nivel septal y anterior.
Figura 2. Resonancia magnética que muestra hipertrofia ventricular izquierda con septo de 31 mm 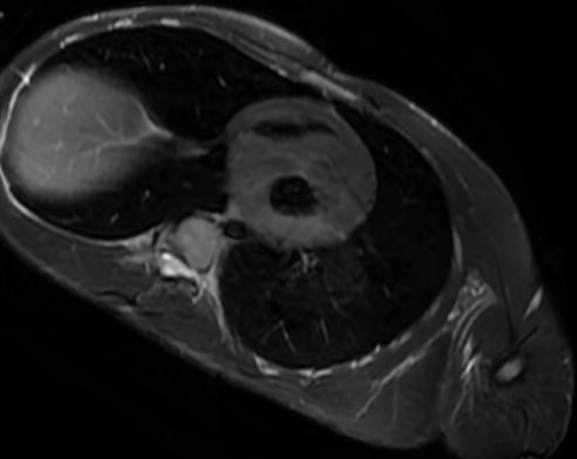

- Estudio genético: portador en homocigosis de mutación en gen TRIM63.

Como antecedentes familiares destaca que es hijo adoptado, con historia familiar desconocida, pero sí se conoce que es procedente de etnia gitana.
Se implanta un desfibrilador automático implantable (DAI) subcutáneo, con buena evolución posterior hasta la fecha.
DISCUSIÓN
La incidencia pediátrica estimada de MCH es de 0,3 a 0,5 casos por 100 000. Es la enfermedad cardiaca genética más prevalente en adultos (1:500 personas, siendo sintomáticos menos de 1:3000)1. La edad de aparición es muy variable. En general, hay mejor pronóstico que en otras miocardiopatías, pero se debe considerar el riesgo de muerte súbita y que algunos subgrupos evolucionan peor2-4.
Las causas más frecuentes de MCH son genéticas y hay síndromes específicos que cursan con MCH.
En la MCH se observan cambios miocárdicos con aumento del grosor y fibrosis, que dan lugar a un ventrículo pequeño y rígido, disfunción diastólica y posteriormente sistólica. La HVI localizada en el tracto de salida del ventrículo izquierdo puede ocasionar obstrucción a la salida de sangre del VI (OTSVI), condicionando la aparición de un soplo, así como anomalías en la función de la válvula mitral. A nivel histológico, se produce una desorganización de miocitos y miofibrillas, fibrosis miocárdica y enfermedad de pequeño vaso, que pueden sentar la base del riesgo arrítmico.
Respecto a la clínica, es frecuente que el niño esté asintomático y que se le esté estudiando por otro motivo. A veces, la presencia de un soplo puede motivar la derivación al especialista. En el 10-15% de los casos la presentación es de insuficiencia cardiaca (IC) o arritmias. Estas últimas pueden debutar como síncope de características cardiogénicas o síncope atípico. Las formas de MCH asociadas a errores congénitos del metabolismo o malformaciones sindrómicas suelen tener un inicio más precoz y más clínica de IC al diagnóstico, siendo la muerte súbita más rara.
El diagnóstico se basa en la confirmación por imagen de la hipertrofia miocárdica. El paciente puede haber presentado síntomas previamente o alteraciones en un ECG o realizarse el estudio por despistaje familiar.
Respecto al ECG, se trata de una prueba accesible desde Atención Primaria, y en más del 90% de los casos se presentan anomalías. Los elementos de sospecha son la hipertrofia por criterios de voltaje, la inversión de la onda T (más específico), las ondas P anchas, las ondas Q patológicas, un intervalo PR corto o preexcitación. El ecocardiograma suele bastar para confirmar el diagnóstico y determina también si hay alteraciones asociadas. La RM cardiaca permite evaluar la presencia de fibrosis, en relación con el riesgo arrítmico. El Holter ECG tiene utilidad en el seguimiento si el niño tiene palpitaciones o presíncopes, y para detectar la aparición de arritmias que confieran mayor riesgo. En la ergometría, se puede evaluar la situación funcional y observar si sucede hipotensión con el esfuerzo, dato de peor pronóstico. En cuanto a las formas metabólicas de niños más pequeños, determinados hallazgos analíticos ayudan a orientar la causa. Y, por último, el estudio genético confirma el origen de la MCH, pero no es necesario encontrar una mutación genética patogénica para que el niño con el fenotipo compatible sea diagnosticado. Respecto a la mutación del gen TRIM63 de este caso, parece que es causa de MCH con un patrón de herencia autosómico recesivo, y se ha propuesto como candidato para el desarrollo de cardiomiopatías (hasta entonces más del 50% de pacientes con MCH no mostraban alteraciones conocidas en estudios genéticos), si bien la evidencia es limitada5.
Los niños con MCH inicialmente pueden no necesitar medicación, sin embargo; el manejo sí puede implicar el implante de un desfibrilador (DAI) que mejore el pronóstico en los grupos de alto riesgo de muerte súbita. Los supervivientes de un evento de este tipo, o que han experimentado una taquiarritmia ventricular con síncope tienen indicación de implante. En el resto de los casos, incluso en ausencia de síntomas, se estratifica el riesgo4,6,7. El DAI reconoce arritmias ventriculares y es capaz de abortarlas. Su implante es similar al de un marcapasos, y el niño debe evitar actividades de contacto que supongan un traumatismo sobre el dispositivo o desplazamiento del cable. La familia debe recibir también información sobre eventuales interacciones electromagnéticas como, por ejemplo, con la RM (aunque muchos DAI son actualmente compatibles) o con el uso de bisturí eléctrico (precisa reprogramación transitoria). Los medicamentos habitualmente empleados son los betabloqueantes cuando hay OTSVI, para intentar reducirla; y en caso de mala tolerancia o contraindicación, los calcioantagonistas. Algunos antiarrítmicos, como la amiodarona, pueden añadirse ante la aparición de arritmias ventriculares. Respecto a las opciones quirúrgicas, estas se plantean en formas graves de OTSVI. En las fases finales de la enfermedad, con insuficiencia cardiaca terminal, está indicado el trasplante cardiaco, en general con buenos resultados8.
CONFLICTO DE INTERESES
La autora declara no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
La autora ha remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
DAI: desfibrilador automático implantable · ECG: electrocardiograma · HVI: hipertrofia ventricular izquierda · IC: insuficiencia cardiaca · Lpm: latidos por minuto · MCH: miocardiopatía hipertrófica · OTSVI: obstrucción al tracto de salida del ventrículo izquierdo · ProBNP: propéptido natriurético cerebral · QTc: intervalo QT corregido · RM: resonancia magnética · VD: ventrículo derecho · VI: ventrículo izquierdo.
BIBLIOGRAFÍA
- Geske JB, Ommen SR, Gersh BJ. Hypertrophic Cardiomyopathy: Clinical Update. JACC Heart Fail. 2018;6(5):364-75. https://doi.org/10.1016/j.jchf.2018.02.010
- Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, et al. Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed During Childhood: Results from a National Population-Based Study. Circulation. 2018;138:29-36. https://doi.org/10.1161/CIRCULATIONAHA.117.028895
- Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020;76:e159-e240. https://doi.org/10.1161/CIR.0000000000000937
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733-79. https://doi.org/10.1093/eurheartj/ehu284
- Salazar-Mendiguchía J, Ochoa JP, Palomino-Doza J, Domínguez F, Díez-López C, Akhtar M, et al. Mutations in TRIM63 cause an autosomal-recessive form of hypertrophic cardiomyopathy. Heart. 2020;106:1342-8. https://doi.org/10.1136/heartjnl-2020-316913
- Norrish G, Ding T, Field E, Ziólkowska l, Olivotto I, Limongelli G, et al. Development of a Novel Risk Prediction Model for Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy (HCM Risk-Kids). JAMA Cardiol. 2019;4:918-27. https://doi.org/10.1001/jamacardio.2019.2861
- Miron A, Lafreniere-Roula M, Steve Fan CP, Armstrong KR, Dragulescu A, Papaz T, et al. A Validated Model for Sudden Cardiac Death Risk Prediction in Pediatric Hypertrophic Cardiomyopathy. Circulation. 2020;142:217-29. https://doi.org/10.1161/CIRCULATIONAHA.120.047235
- Cartón Sánchez AJ, Gutiérrez-Larraya F. Miocardiopatías. Pediatr Integral. 2021;XXV(8):427-36.