
Vol. 27 - Num. 105
Notas clínicas
Síndrome de Sebastian como causa de trombocitopenia
Mercedes Cemeli Canoa, Eva M.ª Jiménez Péreza, Sara Beltrán Garcíaa, Paula Vidal Lanaa, M.ª Jordana Díaz Alonsob, Elena Borque Navarroa
aPediatra. CS Valdespartera. Zaragoza. España.
bEnfermera. CS Valdespartera. Zaragoza. España.
Correspondencia: M Cemeli. Correo electrónico: cano.mcemeli@salud.aragon.es
Cómo citar este artículo: Cemeli Cano M, Jiménez Pérez EM, Beltrán García S, Vidal Lana P, Díaz Alonso MJ, Borque Navarro E. Síndrome de Sebastian como causa de trombocitopenia . Rev Pediatr Aten Primaria. 2025;27:49-54. https://doi.org/10.60147/8db66f2f
Publicado en Internet: 15-01-2025 - Número de visitas: 12492
Resumen
El síndrome de Sebastian es una rara enfermedad relacionada con mutaciones del gen MYH9, con un patrón autosómico dominante donde dicho gen puede dar lugar a diferentes fenotipos en individuos con la misma mutación e incluso dentro de la misma familia. Su importancia radica en la posibilidad de desarrollar insuficiencia renal, hipoacusia y cataratas en edad juvenil o adulta.
Describimos el caso de una niña de 9 años, caucásica, con estudio inicial de trombocitopenia y antecedente familiar de síndrome de Sebastian con estudio genético positivo a la mutación en el gen MYH9.
Destacamos la importancia del diagnóstico precoz de las trombocitopenias de origen hereditario, así como la utilidad del estudio genético para estas enfermedades muy poco frecuentes.
Palabras clave
● Síndrome de Sebastian ● Trastornos relacionados con MYH9 ● TrombocitopeniaINTRODUCCIÓN
El síndrome de Sebastian, también llamado macrotrombocitopenia autosómica dominante con inclusiones leucocitarias, es un raro trastorno genético que se presenta con plaquetas de tamaño mayor al normal y una disminución en el recuento total de plaquetas que altera el funcionamiento adecuado de coagulación.
Está clasificada dentro del grupo heterogéneo de las macrotrombocitopenias, entre las que se encuentran la anomalía de May-Hegglin y los síndromes de Epstein, Alport-like con macrotrombocitopenia, Fechtner y Sebastian) (Tabla 1)1-4. Pueden incluir otros hallazgos, como nefritis hereditaria, sordera neurosensorial progresiva (entre la primera y sexta década de la vida)5,6, inclusiones leucocitarias (cuerpo de Döhle) y cataratas preseniles1. Su tendencia al sangrado es muy variable, por una disminución de los niveles de la expresión del complejo GPIb-V-IX en la superficie de las plaquetas, que implica una alteración de la adhesión y de la agregación plaquetaria.
| Tabla 1. Macrotrombocitopenias relacionadas con el gen MYH9. Signos y síntomas asociados a la alteración plaquetaria8 | ||||
|---|---|---|---|---|
| Enfermedad | Hipoacusia | Cataratas | Nefropatía | Inclusiones leucocitarias |
| Anomalía de May-Hegglin | No | No | No | Sí |
| Síndrome de Sebastian | No | No | No | Sí |
| Síndrome de Fechtner | Sí | Sí | Sí | Sí |
| Síndrome de Epstein | Sí | No | Sí | No |
| Síndrome de Alport-like con macrotrombocitopenia | Sí | Sí | Sí | No |

El trastorno está causado por una mutación autosómica dominante en el gen MYH9, localizado en el brazo largo del cromosoma 22 (22q12.3-q13.2), que codifica para la cadena pesada de la miosina no muscular IIA, NMMHC-IIA. No parece haber diferencias con respecto al sexo, etnia ni lugar geográfico. Hoy en día se considera a dichas alteraciones como un amplio espectro de manifestaciones fenotípicas causadas por mutaciones en el mismo gen7. Desde la clonación del gen se han descrito más de 20 mutaciones distintas en MYH9 en 67 familias no emparentadas y en diferentes partes del mundo. Las macrotrombocitopenias relacionadas con el gen MYH9 se acompañan de una marcada alteración del citoesqueleto de las plaquetas.
El fenotipo de una persona con una variante patogénica en MYH9 a menudo evoluciona con el tiempo. Además, los cinco síndromes no definen todas las manifestaciones posibles derivadas de las variantes patogénicas heterocigotas en MYH9. Por otro lado, los miembros de la misma familia pueden tener diferentes fenotipos y recibir diferentes diagnósticos. Por estas razones, las patologías asociadas a MYH9 han sido propuestas como una nueva entidad nosológica que incluye a todos los individuos con variantes patogénicas heterocigotas en MYH9, independientemente del aspecto de los neutrófilos y el fenotipo clínico. La afectación renal se ha observado en el 30% de los pacientes con mutaciones en el gen, pudiendo progresar a enfermedad renal crónica alrededor de los 30 años1,8,9.
Su incidencia es incierta debido a los limitados casos reportados a nivel mundial. Las formas leves suelen ser un hallazgo incidental, mientras que las graves se diagnostican erróneamente, por lo que su prevalencia quizá es más alta10. Las personas afectadas con este síndrome pueden presentar sangrados leves o más abundantes, e incluso estar asintomáticos (lo más frecuente). El principal riesgo asociado a este síndrome sería la hemorragia durante o después de la cirugía, llegando a precisar transfusión de plaquetas.
El diagnóstico del síndrome de Sebastian se basará en los hallazgos clínicos (historia de hematomas y sangrado excesivo tras medidas de hemostasia)11, junto a las pruebas de laboratorio, donde destacan:
- Plaquetas con volumen plaquetar medio aumentado (VCM) aumentado.
- Trombocitopenia.
- Inclusiones leucocitarias: cuerpos de Döhle (presentes en el 42-84%)12.
El diagnóstico definitivo lo dará el estudio genético, identificándose alteraciones patogénicas en el gen MYH9.
Sus principales diagnósticos diferenciales en la consulta deben ser las trombocitopenias congénitas y adquiridas, entre las que cabe destacar la púrpura trombocitopénica idiopática (PTI).
Debido a sus posibles, aunque poco frecuentes, manifestaciones extrahematológicas, estos pacientes necesitan una evaluación y seguimiento estrecho por un equipo multidisciplinar.
En general, el pronóstico es bueno y no es necesario ningún tratamiento, ya que la mayoría de las hemorragias suelen ser leves y autolimitadas. Se ha de recordar que en el caso de someterse a una intervención quirúrgica mayor puede ser necesaria una transfusión de plaquetas para evitar hemorragias excesivas. Las implicaciones anestésicas son poco conocidas, por el escaso número de casos descritos y porque no se ha encontrado bibliografía sobre su manejo. El diagnóstico prenatal y el diagnóstico preimplantacional son posibles, siempre y cuando se conozca previamente la mutación en el gen MYH9 en el progenitor.
CASO CLÍNICO
Se presenta el caso de una niña de 9 años, caucásica, hija de padre diagnosticado recientemente de síndrome de Sebastian (con mutación del gen MYH9 c,3493C>T), sin otros antecedentes de interés. No ha tenido ingresos ni cirugías previas. Está correctamente vacunada según el calendario vigente. No tiene alergias conocidas. No refiere alteraciones de la función renal, visuales y/o auditivas en el momento actual.
Se remite a consultas de Hematología desde su centro de salud para estudio, dados los antecedentes previamente mencionados en el padre.
Clínicamente, está asintomática desde el punto de vista de diátesis hemorrágica, y no ha tenido la menarquia.
Analíticamente, se observan todos los parámetros bioquímicos sin hallazgos significativos. Desde el punto de vista hematimétrico, se observa una trombocitopenia leve (plaquetas 111 000 x 103/µL), con VPM aumentado (15,2 fL) y presencia de pequeñas inclusiones basófilas de forma semilunar en la parte externa del citoplasma de los neutrófilos. Las pruebas de cribaje de función plaquetar se encuentran dentro de la normalidad.
La presencia de hallazgos analíticos concordantes (trombocitopenia leve con volumen plaquetar elevado y a priori sin alteraciones funcionales) sugerirían el diagnóstico de macrotrombocitopenia familiar (síndrome de Sebastian).
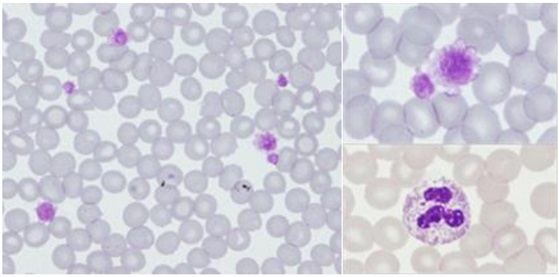
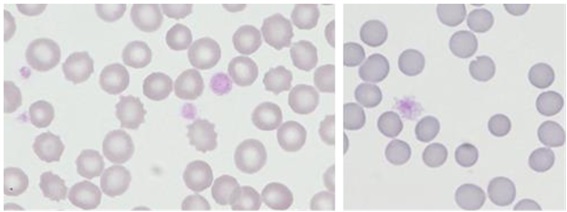
Se enviaron las muestras para la ampliación del estudio molecular y genético al centro de referencia, confirmándose la macrotrombocitopenia moderada tanto en el padre como la hija, con el resto de las series hemoperiféricas dentro de la normalidad (Figuras 1 y 2).
| Figura 1. Anisotrombia marcada con >60% de plaquetas grandes (y dentro de ese porcentaje, un 20% de plaquetas gigantes) |
|---|
 |

| Figura 2. Anisotrombia marcada con 40% de plaquetas grandes (y dentro de ese porcentaje, un 10% de plaquetas gigantes) |
|---|
 |

Además, tanto el padre como la niña presentaron un patrón de expresión de receptores plaquetarios aumentados para la mayoría de glicoproteínas, lo cual es concordante con el tamaño plaquetario aumentado en ambos.
El estudio genético de ambos mostró que son portadores en heterocigosis de la variante en MYH9 c,3493C>T [p.Arg1165Cys], la cual no está presente en la población general, y está clasificada como patogénica según criterios del American College of Medical Genetics and Genomics (ACMG) y ClinVar. Este hallazgo molecular, junto con el fenotipo clínico, confirma el diagnóstico de enfermedad relacionada con MYH9 (MYH9-RD) en ambos pacientes.
DISCUSIÓN
El síndrome de Sebastian forma parte del grupo heterogéneo de enfermedades raras asociadas a mutaciones del gen MYH9, presentando la mayoría desde el nacimiento una macrotrombocitopenia. Los pacientes que padecen alguna de estas enfermedades con mutaciones en dicho gen también desarrollan hipoacusia, cataratas y glomerulopatía, que en algunos casos conduce a la insuficiencia renal; por lo que su diagnóstico precoz y manejo multidisciplinar serán claves para su pronóstico.
Nuestra paciente presentaba cifras previas de plaquetas normales, no siendo hasta la preadolescencia cuando se inicia el estudio de trombocitopenias.
Las causas más frecuentes de trombocitopenia a esta edad7 son las infecciosas (sarampión, varicela, infección por virus de Epstein Barr, citomegalovirus, Helicobacter pylori, parásitos), seguidas de las autoinmunes (púrpura trombocitopénica inmune, lupus, enfermedad inflamatoria intestinal), las inducidas por fármacos (antibióticos, quinidina, ácido valproico, heparina), las inmunodeficiencias, etc.
Es necesario destacar que una misma mutación puede provocar manifestaciones fenotípicas diferentes en individuos distintos e incluso dentro de la misma familia, por lo que se ha sugerido que otros factores podrían mediar en la expresión clínica de la mutación. Un trabajo reciente sugiere que el gen que codifica la fibulina-1, una proteína de la matriz extracelular, ampliamente expresada en la membrana basal de la mayoría de los órganos, podría ser uno de los modificadores de la expresión fenotípica del defecto genético a nivel del gen MYH913.
Por otro lado, Tavira et al., mediante el estudio español RENASTUR, demostraron cierta asociación entre el polimorfismo rs3752462 de MYH9 y el riesgo de desarrollar enfermedad renal crónica14. Apoyando estos resultados, en un estudio de casos y controles de China, se ha descrito la asociación del polimorfismo rs3752462 de MYH9 en pacientes con hipertensión arterial y enfermedad renal crónica15.
En modelos animales con variantes en MYH9 que alteran la estructura del citoesqueleto del podocito, también se ha observado que el estímulo con agentes secundarios como fármacos nefrotóxicos (doxorrubicina y adriamicina), infección por VIH (virus de la inmunodeficiencia humana) e hipertensión arterial favorecen el deterioro de función renal y la aparición de albuminuria16-18.
El diagnóstico de nuestra paciente se apoya también en la detección de la mutación por parte de la rama paterna, siendo heredada al 50% en su descendencia con sintomatología leve.
Por ello, ante la persistencia de una trombocitopenia con el resto de los estudios de primer nivel normales, es de vital importancia descartar causas hereditarias, indagando en los antecedentes familiares y solicitando un estudio genético.
Debido a sus posibles manifestaciones extrahematológicas, estos pacientes necesitan una evaluación y seguimiento multidisciplinar.
CONCLUSIONES
Es importante tener conocimiento sobre estas entidades con mutación del gen MYH9 que, aunque son causa poco frecuente de trombocitopenia, pueden conllevar complicaciones más importantes, como la afectación renal, ocular y de audición.
El pronóstico del síndrome de Sebastian es generalmente bueno, aunque existen algunos polimorfismos en MYH9 que están asociados a mayor riesgo de desarrollar enfermedad renal crónica, destacando la elevada variabilidad genotipo-fenotipo que presentan individuos con la misma mutación.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Contribución de los autores: búsqueda bibliográfica, redacción del manuscrito y enlace con el caso clínico (MCC), búsqueda bibliográfica (EMJP, MJDA y SBG), elaboración de tablas e imágenes (PVL), redacción del caso clínico y revisión final (EN).
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
PTI: púrpura trombocitopénica idiopática · VCM: volumen plaquetar medio aumentado (VCM).
BIBLIOGRAFÍA
- Arrondel C, Vodovar N, Knebelmann B, Grünfeld JP, Gubler MC, Antignac C, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. https://doi.org/10.1681/ASN.V13165
- Girolami A, Vettore S, Bonamigo E, Fabris F. Thrombotic events in MYH9 gene-related autosomal macrothrombocytopenias (old May-Hegglin, Sebastian Fechtner and Epstein syndromes). J Thromb Thrombolysis. 2011;32:474-7. https://doi.org/10.1007/s11239-011-0623-4
- Savoia A, De Rocco D, Pecci A. MYH9 gene mutations associated with bleeding. Platelets. 2017;28:312-5. https://doi.org/10.1080/09537104.2017.1294250
- Rodriguez V, Nichols WL, Charlesworth JE, White JG. Sebastian platelet syndrome: a hereditary macrothrombocytopenia. Mayo Clin Proc. 2003;78:1416-21. https://doi.org/10.4065/78.11.1416
- Verver EJ, Topsakal V, Kunst HP, Huygen PL, Heller PG, Pujol-Moix N, et al. Nonmuscle myosin heavy chain IIA mutation predicts severity and progression of sensorineural hearing loss in patients with MYH9-related disease. Ear Hear. 2016;37:112-20. https://doi.org/10.1097/AUD.0000000000000198
- Aoki T, Kunishima S, Yamashita Y, Minamitani K, Ota S. Macrothrombocytopenia with congenital bilateral cataracts: a phenotype of MYH9 disorder with exon 24 indel mutations. J Pediatr Hematol Oncol. 2018;40:76-8. https://doi.org/10.1097/MPH.0000000000000998
- Merino A. Diagnóstico diferencial de las trombocitopenias. Ed Cont Lab Clín. 2014;17:48-61.
- Otero A, Borrajo MP, Camba MJ, Santos J, Novoa E, Pérez C, et al. Estimated glomerular filtration rate (eGFR), 25(OH) D3, chronic kidney disease (CKD), the MYH9 (myosin heavy chain 9) gene in old and very elderly people. Int Urol Nephrol. 2015;47:1403-8. https://doi.org/10.1007/s11255-015-1041-x
- Cyrus C, Al-Mueilo S, Vatte C, Chathoth S, Li YR, Qutub H, et al. Assessing known chronic kidney disease associated genetic variants in Saudi Arabian populations. BMC Nephrol. 2018;19:88. https://doi.org/10.1186/s12882-018-0890-9
- Pecci A, Panza E, Pujol-Moix N, Klersy C, Di Bari F, Bozzi V, et al. Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9-related disease. Hum Mutat. 2008;29:409-17. https://doi.org/10.1002/humu.20661
- Pecci A, Panza E, De Rocco D, Pujol-Moix N, Girotto G, Podda l, et al. MYH9 related disease: four novel mutations of the tail domain of myosin-9 correlating with a mild clinical phenotype. Eur J Haematol. 2010;84:291-7. https://doi.org/10.1111/j.1600-0609.2009.01398.x
- Noris P, Klersy C, Gresele P, Giona F, Giordano P, Minuz P, et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: a multicentric, real life study. Br J Haematol. 2013;162:112-9. https://doi.org/10.1111/bjh.12349
- Furlano M, Arlandis R, Del Prado M, Novelli S, Crespi J, Bullich G, et al. Nefropatía asociada a mutación del gen MYH9. Nefrologia. 2019;39(2): 133-40. https://doi.org/10.1016/j.nefro.2018.08.008
- Tavira B, Coto E, Gómez J, Tranche S, Miguélez K, Ortega F, et al. Association between a MYH9 polymorphism (rs3752462) and renal function in the Spanish RENASTUR cohort. Gene. 2013;520:73-6. https://doi.org/10.1016/j.gene.2013.02.024
- Liu l, Wang C, Mi Y, Liu D, Li l, Fan J, et al. Association of MYH9 polymorphisms with hypertension in patients with chronic kidney disease in China. Kidney Blood Press Res. 2016;41:956-65. https://doi.org/10.1159/000452597
- Cechova S, Dong F, Chan F, Kelley MJ, Ruiz P, Le TH. MYH9 E1841K mutation augments proteinuria and podocyte injury and migration. J Am Soc Nephrol. 2018;29:155-67. https://doi.org/10.1681/ASN.2015060707
- Johnstone DB, Zhang J, George B, Léon C, Gachet C, Wong H, et al. Podocyte-specific deletion of Myh9 encoding nonmuscle myosin heavy chain 2A predisposes mice to glomerulopathy. Mol Cell Biol. 2011;31:2162-70. https://doi.org/10.1128/MCB.05234-11
- Johnstone DB, Ikizler O, Zhang J, Holzman LB. Background strain and the differential susceptibility of podocyte-specific deletion of Myh9 on murine models of experimental glomerulosclerosis and HIV nephropathy. PLoS One. 2013;8:e67839. https://doi.org/10.1371/journal.pone.0067839