
Vol. 26 - Num. 104
Notas clínicas
Presentación inusual de miastenia gravis juvenil
Vanessa Yareli Solis Poncea, Fernanda Michelle Balderas Martín del Campoaa, Sofía Guadalupe Ocón Garcíab
aMédico cirujano. Universidad Autónoma de Aguascalientes. México.
bPediatra. Instituto Mexicano del Seguro Social. México.
Correspondencia: SG Ocón. Correo electrónico: dra.sofia.ocon@gmail.com
Cómo citar este artículo: Solis Ponce VY, Balderas Martín del Campoa FM, Ocón García SG. Presentación inusual de miastenia gravis juvenil . Rev Pediatr Aten Primaria. 2024;26:393-6. https://doi.org/10.60147/6d751d6e
Publicado en Internet: 13-11-2024 - Número de visitas: 10140
Resumen
La miastenia gravis juvenil es una enfermedad poco frecuente en Pediatría, causada por anticuerpos específicos contra proteínas de la unión neuromuscular, con manifestaciones clínicas puramente oculares o generalizadas que mejoran al reposo. El diagnóstico es clínico, pero la presencia de anticuerpos específicos o anomalías en las conducción nerviosa confirman el diagnóstico. El tratamiento combina sintomáticos, inmunosupresores y timectomía en algunos casos.
Palabras clave
● Mialgia ● Miastenia gravis juvenil ● Síndrome de Wolf-Parkinson-WhiteINTRODUCCIÓN
La miastenia gravis (MG) es una enfermedad causada por anticuerpos específicos contra proteínas postsinápticas de la unión neuromuscular, en más del 90% se trata de anticuerpos contra el receptor de acetilcolina (anti-AChR), aunque también se pueden encontrar anticuerpos contra la quinasa específica de músculo (anti-MuSK) o contra la proteína relacionada con el receptor de lipoproteína 4 (anti-LRP4)1,2. En la población pediátrica se clasifica ─de acuerdo a su edad de aparición─ en miastenia gravis neonatal transitoria (en hijos de madres con MG), síndrome miasténico congénito (por afección génica de la placa neuromuscular) y miastenia gravis juvenil (MGJ)3.
La MGJ se presenta en menores de 18 años con una incidencia de 0,9-8,9/millón de personas por año, con variabilidad étnica y predominio en mujeres. Las manifestaciones clínicas suelen ser puramente oculares, con ptosis y oftalmoplejía, o generalizadas, con dificultad para elevar los brazos, correr, subir escaleras, etc. La sintomatología fluctúa durante el día, pero frecuentemente mejora con reposo. Algunos fenotipos de MGJ puramente ocular pueden desarrollar MGJ generalizada en los primeros 6 meses de inicio de los síntomas4. El diagnóstico es clínico; sin embargo, la detección serológica de anticuerpos anti-AchR, anti-MuSK o anti-LRP4 o la presencia de anomalías en las pruebas neurofisiológicas confirman el diagnóstico5. El abordaje del paciente es multidisciplinario y el tratamiento es una combinación de sintomáticos, como inhibidores de la acetilcolinesterasa (AChEI), inmunosupresores y timectomía en casos apropiados. La tasa de remisión espontánea se presenta entre el 17-55% de los pacientes con MGJ, siendo mayor en niños prepuberales4. En los primeros dos años de la enfermedad se pueden exacerbar los síntomas hasta ocasionar una crisis miasténica que derive en insuficiencia respiratoria y muerte6.
CASO CLÍNICO
Niña de 11 años de edad sin antecedentes heredofamiliares de importancia. Nace por cesárea por placenta previa, sin complicaciones perinatales. Desarrollo psicomotor adecuado. Esquema de vacunación completo para la edad.
Hospitalización a los 5 años de edad por dolor en miembros pélvicos que limita la movilización, disminución de la fuerza, laboratorios con creatina-fosfocinasa (CPK) elevada, manejada con reposo y analgésicos ante sospecha de miositis viral. Posteriormente, presenta cuadros fluctuantes de dolor punzante en piernas después de hacer ejercicio que mejora con el reposo. Hospitalización a los 9 años de edad por dolor intenso en miembros pélvicos que limita la movilización, disminución de la fuerza y sensibilidad desde región lumbar, manejada con analgésicos, con remisión.
Nuevamente, a los 11 años de edad, tras realizar actividad física extenuante presenta dolor en ambos muslos, 8/10 en escala visual análoga, con irradiación a región lumbar; sin respuesta a analgésicos, sensibilidad conservada, fuerza 1/5 en miembro pélvico izquierdo y 3/5 en miembro pélvico derecho según la escala de Daniel´s, reflejos osteotendinosos disminuidos en ambas piernas, reflejos patológicos negativos. Funciones mentales íntegras, nervios craneales sin alteraciones, sin ptosis palpebral, sin disfagia ni disfonía.
Laboratorios con citometría hemática, química sanguínea, transaminasas, electrolitos, pruebas de función hepática, tiempos de coagulación, perfil tiroideo, marcadores de daño tisular y enzimas cardiacas y musculares normales. Radiografía de tórax sin evidencia de masas mediastinales. Resonancia magnética nuclear (RMN) lumbosacra sin alteraciones.
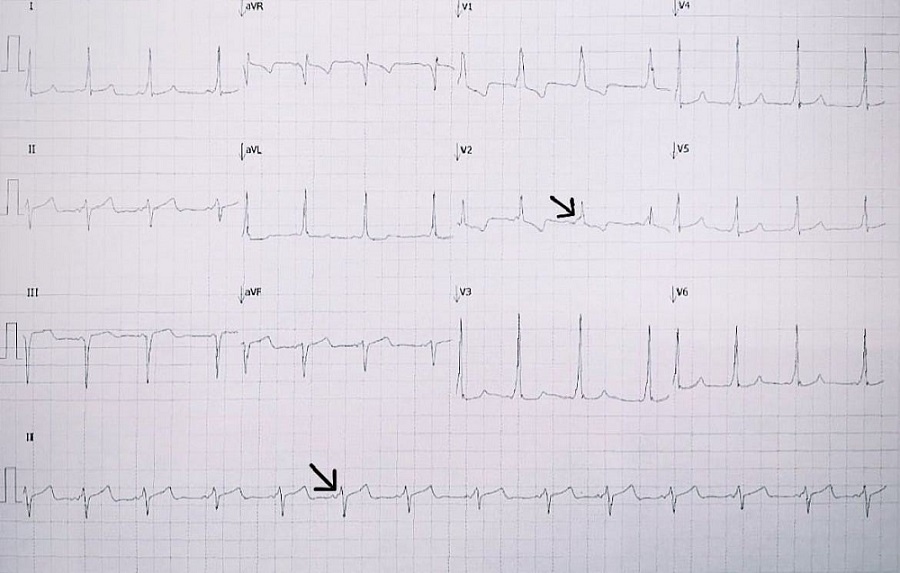
Velocidad de conducción nerviosa normal, electromiografía normal, prueba de estimulación repetitiva con decrementos significativos al estímulo basal, así como después de hacer ejercicio, siendo positivas para enfermedad de placa neuromuscular. La serológica de anticuerpos reporta anti-AChR positivos, el resto negativos. Se diagnostica miastenia gravis juvenil tipo II A, de acuerdo con la clasificación de Osserman (Tabla 1)7. Se inicia tratamiento con prednisona (inmunosupresor) y piridostigmina (AChEI) con buena respuesta al manejo. Como parte del abordaje se realiza electrocardiograma y se detecta síndrome de Wolff-Parkinson-White (WPW) (Figura 1), sin presencia de sintomatología cardiovascular; ecocardiograma sin alteraciones.
| Tabla 1. Clasificación de Osserman de miastenia gravis10 | |
|---|---|
| Tipo | Características |
| I | Miastenia ocular: debilidad de músculos oculares |
| II | Miastenia generalizada leve: debilidad ligera en músculos diferentes a los oculares |
| III | Miastenia generalizada moderada: debilidad moderada en músculos diferentes a los oculares, puede existir debilidad en músculos oculares |
| IV | Miastenia generalizada severa: debilidad severa en músculos diferentes a los oculares, puede existir debilidad en músculos oculares |
| V | Crisis miasténica, intubación con o sin ventilación asistida |
| Las clasificaciones II a IV pueden dividirse en: | |
| A | Mayor compromiso en músculos de extremidades y/o axiales, con afectación menor a músculos orofaríngeos |
| B | Mayor compromiso en músculos orofaríngeos y/o respiratorios, con afectación menor o igual de músculos de extremidades y/o axiales |

| Figura 1. Síndrome de Wolff-Parkinson-White. Las flechas señalan ondas delta de preexitación ventricular |
|---|
 |

DISCUSIÓN
En este caso, el síntoma cardinal de la paciente es dolor muscular. En la primera exacerbación se acompañó de debilidad y elevación de CPK, sugerente de miositis, proceso benigno y autolimitado que aparece tras un cuadro de infección viral aguda en niños8. En la segunda y tercera exacerbaciones, además de dolor y debilidad asimétrica en piernas, se presentó disminución tanto de sensibilidad como de reflejos osteotendinosos, por lo que era prioritario el diagnóstico diferencial con afección medular o lesión radicular9, siendo descartada con RMN. Al reinterrogar nuevamente a la paciente se encontró que la sintomatología fluctuaba durante el día, siendo más intensa tras la realización de ejercicio y con mejoría al reposo; teniendo en cuenta dichas características semiológicas y una vez descartada la lesión medular, el diagnóstico orientó a miastenia gravis juvenil4, corroborándose con las pruebas neurofisiológicas y la presencia de anticuerpos anti-AChR positivos. El hallazgo de WPW asociado a MGJ encontrado en la paciente es poco común, pese a que estos pacientes pueden llegar a presentar en el 58,1% de los casos afecciones cardiacas funcionales, las más frecuentes son alargamiento del intervalo QT o alternancia en la onda T, no así WPW10. La paciente continuó en seguimiento multidisciplinario por Neurología, Cardiología y Pediatría con buena respuesta al tratamiento.
CONCLUSIONES
La MGJ es poco frecuente en Pediatría, La presencia, además, de características peculiares, como en el caso de nuestra paciente, nos obliga a realizar un diagnóstico diferencial amplio, por lo que la anamnesis y la exploración física completa son clave en su abordaje.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Todos los autores han contribuido de forma equivalente en la elaboración del manuscrito publicado.
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
AChEI: inhibidores de la acetilcolinesterasa · Anti-AChR: anticuerpos contra el receptor de acetilcolina · Anti-LRP4: anticuerpos contra la proteína relacionada con el receptor de lipoproteína 4 · Anti-MuSK: anticuerpos contra la quinasa específica de músculo · CPK: creatina-fosfocinasa · MG: miastenia gravis · MGJ: miastenia gravis juvenil · RMN: resonancia magnética nuclear · WPW: síndrome de Wolff-Parkinson-White.
BIBLIOGRAFÍA
- García Estévez DA, Pardo Fernández J. Miastenia gravis. Actualización diagnóstica y terapéutica. Med Clin (Barc). 2023;161(3):119-27. https://doi.org/10.1016/j.medcli.2023.04.006
- Cortés Enríquez OD, Raya Garza LP, Alvarado Vázquez LB, Garza Hinojosa EI, Vázquez Centeno JJ. Miastenia Gravis Neonatal Transitoria: Presentación de un caso y revisión de la literatura. Acta Pediátrica de México [Internet]. 2024;45(5):453-7. https://doi.org/10.18233/apm.v45i5.2751
- Castro Suárez S, Caparó Zamalloa C, Meza Vega M. Actualización en miastenia gravis. Rev Neuropsiquiatr. 2017;80(4). https://doi.org/10.20453/rnp.v80i4.3239
- O’Connell K, Ramdas S, Palace J. Management of juvenile myasthenia gravis. Front Neurol. 2020;11. https://doi.org/10.3389/fneur.2020.00743
- Ge XS, Wei CJ, Dong H, Zhang YH, Bao XH, Wu Y, et al. Juvenile Generalized Myasthenia Gravis With AChR and MuSK Antibody Double Positivity: A Case Report With a Review of the Literature. Front Pediatr. 2022;10(788353):1-6. https://doi.org/3389/fped.2022.788353
- Zalazar GJ, Diaconchuk MA, Martínez CM, Wettstein LG, Milessi ES. Crisis miasténica: ¿predispone la demora en el diagnóstico o el error diagnóstico a su aparición? Neurol Arg. 2017;9(4):243-6. https://doi.org/10.1016/j.neuarg.2017.06.007
- Ropper AH, Samuels MA, Klein JP, Prasad S. Principios de Neurología de Adams y Victor. 11.ª ed. México: McGraw-Hill Interamericana; 2019.
- García Ros M, Núñez Giralda A, Delgado Fuentes E. Miositis viral aguda: a propósito de ocho casos. Rev Pediatr Aten Primaria. 2017;19:363-5.
- Ariza Jiménez AB, Moreno Muñoz G, Martínez Antón J, Ledesma Albarrán JM. Diagnóstico diferencial de la debilidad muscular. Form Act Pediatr Aten Prim. 2013;6(2):118-40.
- Orea Tejeda A, Oseguera Moguel J, Gómez M, Narváez David R, Castillo Martínez l, Asensio Lafuente E, et al. Descripción del electrocardiograma en reposo de una serie de pacientes con miastenia gravis. Rev Invest Clin. 2003;55(3):270-5.