
Vol. 26 - Num. 103
Notas clínicas
Un caso de exoftalmos progresivo en una niña de 5 años
Marta Martín Gonzáleza, Ana Isabel Pérez Hernándezb, Álvaro Pineda Torcuatoc
aMédico de Familia. CS Torrelodones. Madrid. España.
bPediatra. CS Torrelodones. Madrid. España.
cMédico de Familia. Hospital Universitario Puerta de Hierro. Servicio de Urgencias. Madrid. España.
Correspondencia: M Martín. Correo electrónico: apinedatorcuato@gmail.com
Cómo citar este artículo: Martín González M, Pérez Hernández AI, Pineda Torcuato A. Un caso de exoftalmos progresivo en una niña de 5 años . Rev Pediatr Aten Primaria. 2024;26:301-4. https://doi.org/10.60147/695146e2
Publicado en Internet: 30-09-2024 - Número de visitas: 6715
Resumen
El sarcoma fusocelular es un tumor poco frecuente, con escasa presentación en la literatura. Los tumores de partes blandas constituyen un grupo heterogéneo, precisando un abordaje multidisciplinar.
Escolar de 5 años que acude a su pediatra por exoftalmos. Su madre refiere un cuadro progresivo de proptosis en el ojo izquierdo de 6 meses de evolución. No hay pérdida de agudeza visual, pero en las últimas semanas se queja de dolor intermitente con los movimientos oculares. A la exploración, proptosis del ojo izquierdo con lagrimeo intermitente y lesión vascular en porción temporal del mismo ojo. Presenta restricción de los movimientos del ojo izquierdo. Se deriva a urgencias hospitalarias, donde realizan un TAC craneal, objetivándose una lesión retrorbitaria izquierda de 28 x 14 mm, unilateral, que condiciona proptosis, efecto de masa sobre estructuras retroculares y pérdida de esfericidad ocular. Ante los hallazgos en las pruebas de imagen, se decide intervención quirúrgica por medio de craneotomía frontotemporal, recibiendo posteriormente tratamiento complementario con radioterapia local. Ante negatividad de estudios de patología molecular, se interpretó finalmente el resultado como sarcoma “tipo adulto” fusocelular indiferenciado.
Aunque el sarcoma fusocelular indiferenciado es una patología poco frecuente, debe incluirse en el diagnóstico diferencial de las patologías retrorbitarias. El objetivo de este caso es repasar las principales características del sarcoma fusocelular, dada su rápida y agresiva forma de presentación, en un tumor poco frecuente.
Palabras clave
● Exoftalmos ● Proptosis ● SarcomaINTRODUCCIÓN
El sarcoma fusocelular indiferenciado es un tumor poco frecuente, con escasa presentación en la literatura. Los sarcomas de partes blandas constituyen un grupo heterogéneo de tumores que precisan tratamiento multidisciplinario1.
El objetivo es repasar el sarcoma fusocelular que, pese a ser poco frecuente, presenta una evolución rápida y agresiva.
CASO CLÍNICO
Escolar de 5 años, sin antecedentes personales de interés, que acude a consulta en el centro de salud por exoftalmos. La madre refiere un cuadro de proptosis progresiva en el ojo izquierdo de 6 meses de evolución. Ha recibido diferentes tratamientos ante diversas sospechas diagnósticas, sin respuesta clínica. No refiere pérdida de agudeza visual, pero en la última semana ha referido dolor intermitente asociado a la movilidad ocular. A la exploración física destaca proptosis del ojo izquierdo con lagrimeo intermitente y lesión vascular en porción temporal del mismo ojo. No hay diplopía. Pupilas isocóricas normorreactivas. Movimientos oculares externos (MOE) conservados, aunque presenta restricción de los movimientos del ojo izquierdo. No se palpan adenopatías significativas. No hay signos de focalidad neurológica; el resto de la exploración es normal.
Es derivada a urgencias hospitalarias, donde se realiza TAC craneal sin contraste intravenoso que objetiva una lesión retrorbitaria izquierda de 28 x 14 mm, unilateral, aparentemente extraconal (Figuras 1 y 2), que condiciona proptosis, efecto de masa sobre estructuras retroculares y pérdida de esfericidad ocular; se cosidera como primera posibilidad diagnóstica rabdomiosarcoma vs. lesión vasculogénica (hemangioma infantil o malformación venolinfática).
| Figura 1. TAC sin contraste: cortes axial, sagital y frontal |
|---|
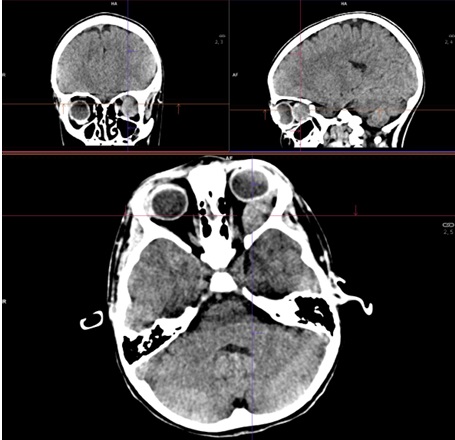 |

| Figura 2. TAC sin contraste: corte sagital |
|---|
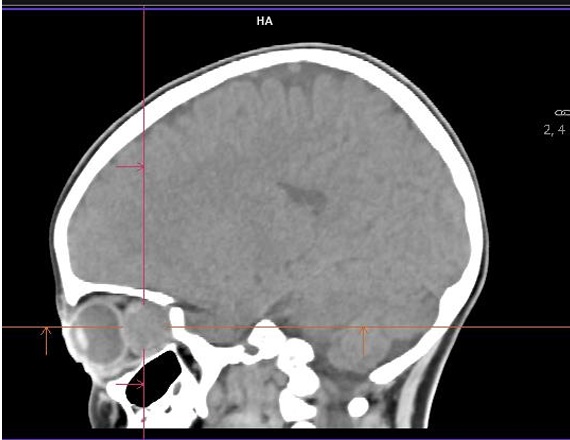 |

El estudio analítico inicial muestra hemograma y coagulación normales; en bioquímica destaca LDH 348 UI/l (niveles normales: 110-295). Se realizan una radiografía simple de tórax y una ecografía abdominal normales, y resonancia magnética nuclear (RMN) cerebral con hallazgos de masa orbitaria intra/extraconal con extensión periorbitaria y deformidad de pared lateral secundaria.
Ante los hallazgos en las pruebas de imagen, se decide intervención quirúrgica por medio de craneotomía frontotemporal, recibiendo posteriormente tratamiento complementario con radioterapia local.
La anatomía patológica mostró proliferación fusocelular fibroblástica con moderada atipia y aisladas figuras de mitosis. Siendo el fibrosarcoma infantil la primera posibilidad diagnóstica, se realizaron estudios de patología molecular (ETV y NTRK, entre otros) que resultaron negativos.
Ante negatividad de estudios de patología molecular, se interpretó finalmente el resultado como sarcoma “tipo adulto” fusocelular indiferenciado.
Los tumores malignos en la edad pediátrica son poco frecuentes y requieren en su mayoría de estudio anatomopatológico para llegar a un diagnóstico definitivo1,2.
Los sarcomas de partes blandas se clasifican en rabdomiosarcomas y no rabdomiosarcomas.
Por una parte, los rabdomiosarcomas, representan el 5% de los cánceres infantiles3. Tienen un pico de incidencia en la primera infancia y uno segundo en la adolescencia, siendo su localización más frecuente: cabeza y cuello, genitourinarios, extremidades y órbita2.
El rabdomiosarcoma es el tumor orbitario maligno más prevalente en la infancia. Su incidencia en España se desconoce3.
El rabdomiosarcoma primario orbitario se presenta en la primera década de la vida, con tasa hombre/mujer 5:3. Suele manifestarse como una proptosis rápidamente progresiva, unilateral. En cuanto a la extensión tumoral: erosiones óseas, invasión a senos paranasales e invasión intracraneal ocurren entre un 30 y un 40% de los casos; las metástasis se dan por vía hemática a pulmón y hueso3.
Por otra parte, los sarcomas no rabdomiosarcomas se presentan con mayor incidencia en niños más mayores y en la adolescencia2. Los más frecuentes son el sarcoma sinovial, el tumor maligno de la vaina nerviosa, el fibrosarcoma infantil y los sarcomas vasculares2.
Pueden surgir en cualquier parte del organismo y suelen manifestarse como masas sólidas asintomáticas; en algunos casos producen síntomas por invasión local en estructuras anatómicas adyacentes. Los síntomas sistémicos son poco frecuentes4.
Los sarcomas fusocelulares se engloban dentro del grupo de los sarcomas no rabdomiosarcomatosos y no son habituales en la infancia.
Existe un pequeño número de casos representados en la literatura, de los cuales algunos se han descrito asociados a síndromes malformativos, como el síndrome de Mafucci5.
Los sarcomas fusocelulares son tumores de agresividad intermedia, derivados de células mesenquimales6. Se diferencian en función del tipo celular que constituye dicho patrón: células fibroblásticas, miofibroblásticas, endoteliales, músculo liso o células mesenquimales indiferenciadas, entre otras6. Para tipificar los distintos tipos celulares se emplean paneles inmunohistoquímicos6.
En el caso del sarcoma indiferenciado, el diagnóstico es de exclusión, ya que no presenta una morfología específica7.
En el diagnóstico diferencial de lesiones retrorbitarias deben incluirse también los tumores o lesiones vasculares, como hemangiomas o malformaciones venolinfáticas; la patología infecciosa, como las celulitis; la patología inflamatoria, como la oftalmopatía tiroidea o el pseudotumor inflamatorio; y las anomalías congénitas3.
El hemangioma es la tumoración benigna de crecimiento lentamente progresivo más frecuente. Se caracteriza por la presencia de sinusoides con contenido hemático sin flujo arterial o venoso aparente. Suele manifestarse como proptosis o exoftalmos de largo tiempo de evolución sin otra sintomatología asociada. El tratamiento puede ser conservador con betabloqueantes o quirúrgico8.
Por otro lado, la malformación venolinfática más prevalente (después de anomalías congénitas del desarrollo) es la malformación cavernomatosa venosa9.
CONCLUSIÓN
El sarcoma fusocelular indiferenciado debe incluirse en el diagnóstico diferencial de lesión retrorbitaria, a pesar de su escasa frecuencia. Puede presentarse como proptosis rápidamente progresiva con o sin otra sintomatología asociada.
Para un correcto diagnóstico y tratamiento son necesarias tanto pruebas de imagen como anatomopatológicas con uso de paneles inmunohistoquímicos de cara a un tratamiento multidisciplinar.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Todos los autores han contribuido de forma equivalente en la elaboración del manuscrito publicado.
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
BIBLIOGRAFÍA
- García del Muro X, Martín J, Maurel J, Cubedo R, Bagué S, De Álava E, et al.; Grupo Español de Investigación en Sarcomas (GEIS). Guía de práctica clínica en los sarcomas de partes blandas [Soft tissue sarcomas: clinical practice guidelines]. Med Clin (Barc). 2011;136(9):408.e1-408.e18. https://doi.org/10.1016/j.medcli.2011.02.004
- Criterios revisados, acordados por el Consejo Interterritorial, que deben cumplir los CSUR para ser designados. En: Ministerio de Sanidad [en línea] [consultado el 14/12/2022]. Disponible en www.sanidad.gob.es/gl/profesionales/CentrosDeReferencia/docs/Fesp/Fesp57r.pdf
- Jiménez Morales ML, Gómez Garza G, Criales Cortés JL, Mora Tiscareño MA. Patología orbitaria en la población pediátrica: revisión de hallazgos mediante resonancia magnética. Anales de Radiología México 2015;14:191-208.
- Tratamiento del sarcoma de tejido blando infantil (PDQ®). En: Instituto Nacional del Cáncer [en línea] [consultado el 14/12/2022]. Disponible en www.cancer.gov/espanol/tipos/sarcoma-de-tejido-blando/pro/tratamiento-tejido-blando-infantil-pdq
- García Ortega DY, Clara Altamirano MA, Álvarez Cano A, Partida Nava GV, Martínez Said H, Caro Sánchez CHS, et al. Doble primario sincrónico: condrosarcoma y sarcoma fusocelular de alto grado asociado a síndrome de Maffucci [Synchronous primary double: condrosarcoma and sarcoma high grade fusocelular associated with Maffucci syndrome]. Acta Ortop Mex. 2019;33(5):325-8.
- Manual de procedimientos en los registros hospitalarios de tumores. En: Sociedad española de anatomía patológica [en línea] [consultado el 14/12/2022]. Disponible en www.seap.es/images/stories/recursos/manual-de-procedimientos-en-los-registros-hospitalarios-de-tumores-2007/Manual_procedimientos_RHT.pdf
- Muñoz Gallego A, Mencía Gutiérrez E, Cámara Jurado M, Gallego Gallego MS, Gutiérrez Díaz E. Sarcoma pleomorfo indiferenciado de alto grado orbitario: a propósito de un caso. Arch Soc Esp Oftalmol. 2014;89(10):425-7.
- Barros Centeno MF, Mansilla C. Patología vascular órbito-palpebral en niños: una serie de casos. Oftalmol Clin Exp. 2022;15(2):e178-e184.
- Aliaga A, Palavecino T, Espinoza R, Dellien H. Malformación cavernomatosa: Revisión de una patología clásica. Rev Chil Radiol. 2013;19(3):117-24. http://dx.doi.org/10.4067/S0717-93082013000300006