
Vol. 26 - Num. 101
Notas clínicas
Malformación de Arnold Chiari
M.ª Tatiana Fernández Garridoa, Jorge Lévano Vásquezb, M.ª Mercedes Santos Herreroa, Jesús M.ª Pascual Pérezb, Anny Vanessa Martínez Báezc
aMédico de Familia y médico puericultor. CS Los Pintores. Parla. Madrid. España
bPediatra. CS Los Pintores. Parla. Madrid. España.
cMédico de Familia. CS Los Pintores. Parla. Madrid. España
Correspondencia: MT Fernández. Correo electrónico: tatiana.fernandez@salud.madrid.org
Cómo citar este artículo: Fernández Garrido MT, Lévano Vásquez J, Santos Herrero MM, Pascual Pérez JM, Martínez Báez AV. Malformación de Arnold Chiari . Rev Pediatr Aten Primaria. 2024;26:59-63. https://doi.org/10.60147/0fd48905
Publicado en Internet: 05-03-2024 - Número de visitas: 23756
Resumen
La malformación de Arnold Chiari es una enfermedad rara que consiste en una alteración anatómica de la base del cráneo, en la que se produce herniación del cerebelo y del tronco del encéfalo a través del foramen magnum hasta el canal cervical. Muchos niños afectados nunca llegan a tener síntomas. En el caso de que cause síntomas, estos no suelen aparecer hasta la infancia tardía o adolescencia. Presentamos el caso clínico de un paciente de 2 años con malformación de Arnold Chiari tipo I.
Palabras clave
● Cefalea ● Malformación de Arnold Chiari ● Síndrome de apneas del sueñoINTRODUCCIÓN
La malformación de Chiari (MC) es una enfermedad rara congénita, que consiste en una alteración anatómica de la base del cráneo, en la que se produce herniación del cerebelo y tronco del encéfalo a través del foramen magnum hasta el canal cervical1.
La MC tipo I (MC-I) es la forma más frecuente. Se desconoce su verdadera prevalencia, siendo del 1 al 3,6% en estudios de resonancia magnética nuclear (RMN) desde su generalización2.
La MC-I suele ser asintomática en niños pequeños. Las manifestaciones clínicas son muy variables y dependen del complejo malformativo asociado y de la existencia o no de cavidades siringomiélicas e hidrocefalia en el momento del diagnóstico. Los síntomas más frecuentes son la cefalea (que se localizan en región occipito-cervical y suele aumentar cuando se realizan maniobras de Valsalva), cervicalgia y mareo. También pueden presentar disfagia, ataxia, alteraciones sensitivas y/o motoras, alteraciones del sueño y trastornos respiratorios nocturnos3,4.
El diagnóstico se realiza mediante RMN, cuando una o ambas amígdalas cerebelosas se desplazan ≥5 mm por debajo del foramen magnum5,6.
En cuanto a su manejo, los pacientes asintomáticos con un diagnóstico incidental de MC-I sin siringomielia ni obstrucción del flujo de líquido cefalorraquídeo (LCR) pueden tratarse de manera conservadora con vigilancia clínica y RMN periódica de control. Por otra parte, aquellos sintomáticos (con cefalea occipital, parálisis de nervios craneales inferiores, síntomas cerebelosos o secundarios a siringomielia) deben ser derivados para valorar indicación quirúrgica (descompresión del foramen magnum)7,8.
CASO CLÍNICO
Presentamos el caso clínico de un paciente de 2 años que acude a la consulta de pediatría de Atención Primaria por ronquido y pausas de apnea durante el sueño de meses de evolución, así como episodios puntuales de cefalea occipital. Se deriva al servicio de otorrinolaringología (ORL). En la exploración física presenta hipertrofia amigdalar grado IV, hipertrofia adenoidea grado III y otitis media serosa bilateral. La pulsioximetría nocturna domiciliaria evidencia un síndrome de apnea hipopnea del sueño (SAHS) leve-moderado. Se programa adenoamigdalectomía y colocación de drenajes transtimpánicos bilaterales sin incidencias.
Tres meses después de la cirugía, consulta de nuevo por persistencia del ronquido y apneas del sueño, así como empeoramiento de la cefalea occipital, más intensa y constante, precisando la toma de analgésicos a diario.
Desde la urgencia hospitalaria se solicita RMN preferente (Figuras 1 y 2), que objetiva herniación de amígdalas cerebelosas de aproximadamente 23 mm (hasta la altura de C2-C3), compatible con MC-I. Además, asocia leve retropulsión de la odontoides que produce compresión del cordón medular, pero sin cambios de señal que sugieran mielopatía en el momento actual.
| Figura 1. Corte sagital de resonancia magnética nuclear donde se objetiva herniación de amígdalas cerebelosas |
|---|
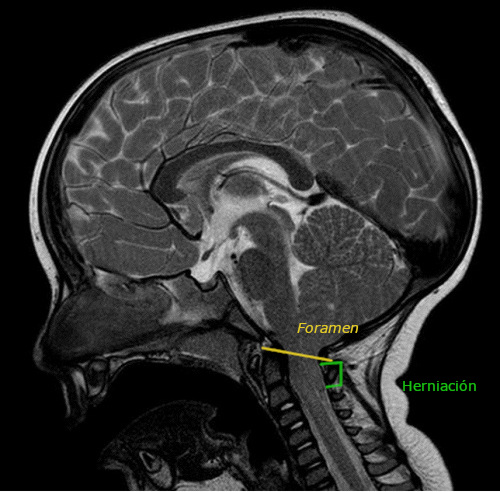 |
| Figura 2. Corte coronal de resonancia magnética nuclear donde se objetiva herniación de amígdalas cerebelosas |
|---|
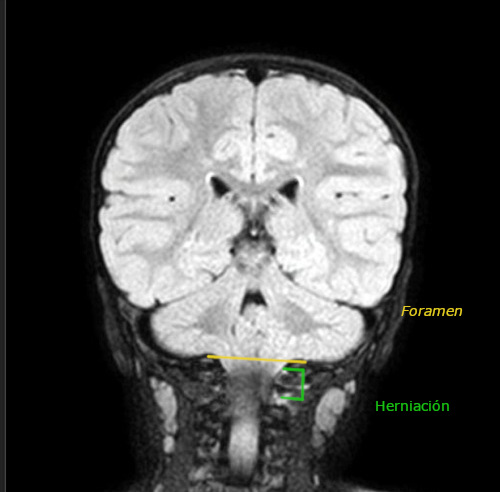 |
Se interviene quirúrgicamente, realizando descompresión suboccipital, exéresis del arco posterior de C1 y coagulación de amígdalas cerebelosas, con buena evolución posquirúrgica.
En los controles sucesivos presenta mejoría clínica y radiológica (RMN) sin complicaciones ni recaídas. Y se normaliza la polisomnografía, sin presentar SAHS.
DISCUSIÓN
La MC es una enfermedad rara, generalmente congénita, que consiste en una alteración anatómica de la base del cráneo en la que se produce herniación del cerebelo y tronco del encéfalo a través del foramen magnum hasta el canal cervical.
Se clasifica en cinco subtipos:
- MC-0: existe alteración de la hemodinámica del líquido cefalorraquídeo (LCR) a nivel del foramen magnum. Los pacientes tienen siringomielia sin datos de herniación amigdalar.
- MC-I (la más frecuente): herniación caudal de las amígdalas cerebelosas mayor de 5 mm por debajo del foramen magnum. No suele acompañarse de descenso del tronco del encéfalo, cuarto ventrículo ni hidrocefalia.
- MC-II: herniación caudal a través del foramen magnum del vermis cerebeloso, tronco del encéfalo y cuarto ventrículo. Se asocia con mielomeningocele e hidrocefalia.
- MC-III: consiste en una encefalocele occipital con parte de las anomalías intracraneales asociadas a Chiari II.
- MC-IV: aplasia o hipoplasia del cerebelo asociada con aplasia de la tienda del cerebelo.
Los síntomas suelen ser de inicio insidioso y curso progresivo. Existe mucha variabilidad clínica entre los pacientes (algunos asintomáticos y otros con manifestaciones clínicas inespecíficas o signos neurológicos graves).
La sintomatología puede ser fluctuante, con periodos de agudización y de remisión.
- Síntomas secundarios a alteración de la dinámica del LCR:
- La cefalea suboccipital es el síntoma más frecuente, de localización occipital, opresiva, y aumenta con maniobras de Valsalva. Puede tener características inespecíficas o mostrar un patrón tensional.
- La cervicalgia es muy frecuente, se caracteriza por carecer de distribución radicular y estar acompañada de molestias continuas, urentes y profundas, localizada en los hombros, nuca, pecho y extremidades superiores, que típicamente aumenta con maniobras de Valsalva.
- Vértigos, sobre todo posicionales o desencadenados por el movimiento de la cabeza.
- Síntomas por compresión de la médula o el bulbo raquídeo:
- Debilidad y espasticidad en las cuatro extremidades.
- Alteraciones sensitivas en extremidades superiores, sobre todo parestesias.
- Apneas.
- Disartria, disfonía o disfagia.
- Síndrome cerebeloso: refieren inestabilidad y dismetría en la coordinación de las extremidades.
- Síndrome centromedular: en los casos asociados a siringomielia. La clínica típica consiste en debilidad segmentaria y atrofia de las manos, con arreflexia e hipoestesia disociada suspendida en tronco o extremidades9.
Existen numerosos estudios sobre los trastornos respiratorios durante el sueño en pacientes con MC-I. La mayor parte de estos han objetivado alteraciones respiratorias de características centrales, pero también se han descrito obstructivas y mixtas. Pueden ser el primer y único síntoma tanto en edad adulta como pediátrica, siendo en esta última en la que aparecen predominantemente de tipo central4.
El diagnóstico del MC-I (tanto en pacientes sintomáticos como asintomáticos) se realiza mediante técnicas de neuroimagen, siendo de elección la RMN. Una vez hecho el diagnóstico de MC-I, el paciente debe ser sometido a estudio del sueño y potenciales evocados, con el fin de valorar el grado de afectación y severidad de la enfermedad y las anomalías asociadas9.
En pacientes sintomáticos, se debe considerar el tratamiento quirúrgico. Todas las técnicas tienen en común la descompresión del foramen magnum9. Esta ha demostrado ser efectiva para los síntomas neurológicos, pero también se obtiene una mejoría de los respiratorios según algunos estudios4. Contribuye fundamentalmente a mejorar los parámetros respiratorios centrales, mientras que en los obstructivos se han encontrado resultados dispares: sin cambios, mejoría parcial o reversión completa de las alteraciones prequirúrgicas.
En nuestro caso, la forma de presentación clínica con apnea del sueño, la corta edad del paciente y la presencia de hipertrofia adenoamigdalar, junto con el estudio de pulsioximetría nocturna con diagnóstico de SAHS leve-moderado, condujo a realizar cirugía ORL. La persistencia de apneas del sueño tras la misma sugiere que no solo eran obstructivas, sino mixtas (como posiblemente se hubiera evidenciado si se hubiera realizado una polisomnografía al principio del estudio). Y en ese caso, ante sospecha de alteración en la base del cráneo, se habría solicitado antes una RMN, favoreciendo una intervención más precoz, incluso antes de comenzar con la cefalea occipital (signo de alarma ante la corta edad del niño). Tras la descompresión del foramen magnum el paciente presentó mejoría clínica, quedando asintomático y con estudio de polisomnografía negativa para SAHS.
CONCLUSIÓN
Aunque la MC-I sea una enfermedad rara y la mayoría de veces asintomática, algunos pacientes presentarán clínica y precisarán tratamiento quirúrgico, por lo que es importante conocer la existencia de esta malformación y pensar en ella. Aunque las formas sintomáticas son más frecuentes en niños mayores y adolescentes, se debe tener en cuenta incluso en más pequeños. El pediatra de Atención Primaria tiene un papel clave gracias a la longitudinalidad, asegurando la continuidad asistencial entre niveles. Los síntomas suelen ser de inicio insidioso y curso progresivo, pudiendo implicar a diversas especialidades (en nuestro caso, ORL, urgencias hospitalarias, neuropediatría y neurocirugía). Ante una sospecha diagnóstica, se realizará la derivación oportuna a especializada para completar el estudio y poder realizar un diagnóstico y manejo lo más precoz posible, evitando posibles complicaciones asociadas y/o intervenciones quirúrgicas más complejas.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Todos los autores han contribuido de forma equivalente en la elaboración del manuscrito publicado. Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
LCR: líquido cefalorraquídeo · MC: malformación de Chiari · ORL: otorrinolaringología · RMN: resonancia magnética nuclear · SAHS: síndrome de apnea hipertrófica del sueño.
BIBLIOGRAFÍA
- Sarnat HB. Disorders of segmentation of the neural tube: Chiari malformations. Handb Clin Neurol. 2008;87:89-103. https://doi.org/10.1016/S0072-9752(07)87006-0
- Speer MC, Enterline DS, Mehltretter l, Hammock P, Joseph J, Dickerson M, et al. Review article: Chiari Type I malformation with or without syringomyelia: prevalence and genetics. J Genet Couns. 2003;12:297-311. https://doi.org/10.1023/A:1023948921381
- Milhorat TH, Chou MW, Trinidad EM, Kula RW, Mandell M, Wolpert C, et al. Chiari I malformation redefined: clinical and radiographic findings for 364 symptomatic patients. 1999;44:1005-17. https://doi.org/10.1097/00006123-199905000-00042
- Ferré Masó A, Poca MA, De la Calzada MD, Solana E, Romero Tomás O, Sahuquillo J. Alteraciones del sueño, un síndrome olvidado en los pacientes con malformación de Chiari tipo I. Neurología. 2014;29(5):294-304. https://doi.org/10.1016/j.nrl.2011.01.008
- Caldarelli M, Di Rocco C. Diagnosis of Chiari I malformation and related syringomyelia: radiological and neurophysiological studies. Child’s Nerv Syst. 2004;20(5):332-5. https://doi.org/10.1007/s00381-003-0880-4
- Wu YW, Chin CT, Chan KM, Barkovich AJ, Ferriero DM. Pediatric Chiari I malformations. Do clinical and radiologic features correlate? Neurology. 1999;53(6):1271. https://doi.org/10.1212/WNL.53.6.1271
- Massimi l, Peretta P, Erbetta A, Solari A, Farinotti M, Ciaramitaro P, et al. Diagnosis and treatment of Chiari malformation type 1 in children: the International Consensus Document. Neurol Sci. 2022;43(2):1311-26. https://doi.org/10.1007/s10072-021-05317-9
- Leon TJ, Kuhn EN, Arynchyna AA, Smith BP, Tubbs RS, Johnston JM, et al. Patients with “benign” Chiari I malformations require surgical decompression at a low rate. JNS.2019;23(4):498-506. https://doi.org/10.3171/2018.10.PEDS18407
- Amado ME, Avellaneda A, Barrón J, Chesa E, De la Cruz J, Escribano M, et al. Malformaciones de la unión craneocervical (Chiari tipo I y siringomielia). Documento de consenso. Editorial Médica AWWE; 2010 [en línea] [consultado el 04/03/2024]. Disponible en www.sen.es/pdf/2010/Consenso_Chiari_2010.pdf