
Vol. 25 - Num. 99
Notas clínicas
Máculas hiperpigmentadas y prolapso rectal, ¿qué debemos sospechar?
Pilar Ferrer Santosa, Diana Clavero Chuecaa, Carmen Navascués Cajalb, Irene Celiméndiz Ferrándezb, Manuel Lavilla Jiménezc
aPediatra. CS Tarazona. Zaragoza. España.
bEnfermera. CS Tarazona. Zaragoza. España.
cMédico de familia. CS Tarazona. Zaragoza. España.
Correspondencia: P Ferrer . Correo electrónico: pilarferrersantos@gmail.com
Cómo citar este artículo: Ferrer Santos P, Clavero Chueca D, Navascués Cajal C, Celiméndiz Ferrández I, Lavilla Jiménez M. Máculas hiperpigmentadas y prolapso rectal, ¿qué debemos sospechar? . Rev Pediatr Aten Primaria. 2023;25:273-6. https://doi.org/10.60147/eb5053f7
Publicado en Internet: 15-09-2023 - Número de visitas: 7944
Resumen
El síndrome de Peutz-Jeghers (SPJ) es un síndrome autosómico dominante con una incidencia de 1 de cada 200 000 nacidos vivos. Las manifestaciones clínicas más frecuentes son las máculas hiperpigmentadas típicamente localizadas en la mucosa oral y la presencia de pólipos en el tracto gastrointestinal.
A diferencia de la edad adulta, en Pediatría es excepcional el desarrollo de patología tumoral maligna. Sin embargo, en la edad pediátrica hay que tener un elevado índice de sospecha ante un paciente con diagnóstico de SPJ que presenta dolor abdominal compatible con una invaginación intestinal, ya que esta complicación es relativamente frecuente y precisa tratamiento quirúrgico urgente. Una vez realizado el diagnóstico de esta enfermedad, se deberán llevar a cabo controles periódicos mediante endoscopias a partir de los ocho años de edad.
Palabras clave
● Hiperpigmentación cutánea ● Poliposis ● Prolapso rectal ● Síndrome Peutz-JeghersINTRODUCCIÓN
El síndrome de Peutz-Jeghers (SPJ) es un síndrome autosómico dominante con una incidencia baja, pero que precisa un manejo y seguimiento muy específicos para evitar el desarrollo de graves complicaciones a lo largo de la vida. Por ello debemos conocer la asociación típica entre hiperpigmentación cutánea y pólipos gastrointestinales para poder llevar a cabo un diagnóstico precoz de esta patología.
En la edad pediátrica las máculas hiperpigmentadas típicamente localizadas en la zona bucal suelen ser la manifestación inicial. Ante la sospecha de esta entidad, se deberá confirmar mediante estudio genético (gen STK11). Una vez confirmado el SPJ, se realizarán controles endoscópicos periódicos a partir de los 8 años, o antes si aparecen síntomas gastrointestinales. La principal complicación en la edad pediátrica es la invaginación intestinal, que precisa tratamiento quirúrgico precoz. Estos pacientes también pueden debutar con una anemia crónica o prolapso rectal. Este síndrome está asociado al desarrollo de múltiples cánceres en edad adulta, por lo que precisarán seguimiento a lo largo de toda la vida1.
CASO CLÍNICO
Paciente de 5 años que acude por primera vez a la consulta del centro de salud para control tras haber sufrido un prolapso rectal reducido manualmente en un centro hospitalario el día anterior.
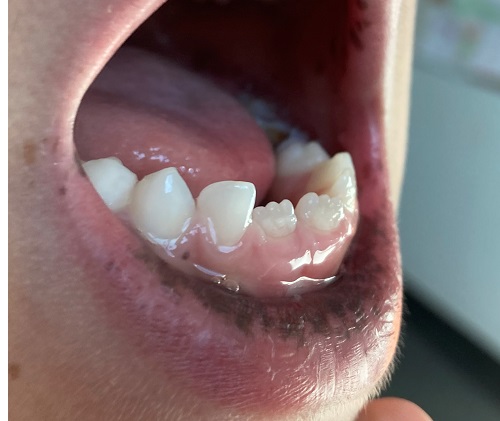
Nacido y controlado hasta entonces en Venezuela. Al realizar la exploración física completa, llaman la atención múltiples máculas hiperpigmentadas localizadas en mucosa yugal, labios, encías y conjuntivas (Fig. 1). El resto de la exploración física es normal. Se consulta a la madre sobre la evolución de estas lesiones y comenta que las tiene desde siempre y le habían informado de que se trataba de máculas benignas asociadas a su fototipo de piel.
| Figura 1. Síndrome de Peutz-Jeghers: hiperpigmentación oral en paciente de 5 años |
|---|
 |
Se interroga sobre antecedentes médicos y quirúrgicos en su país de origen y la madre refiere que tuvo un embarazo controlado, con desarrollo perinatal normal e introducción de la alimentación complementaria sin incidencias. Al año de vida, presentó un prolapso rectal en domicilio, por lo que acudió a un centro hospitalario donde se redujo manualmente y se procedió a la extirpación de 3 pólipos rectales. Desconoce si se analizaron en anatomía patológica y no aporta informes.
Niega antecedentes familiares de patologías digestivas y de lesiones cutáneas similares.
Dada la asociación de los pólipos y las máculas hiperpigmentadas, se remite a consulta de Cirugía y Gastroenterología Pediátrica para descartar SPJ e iniciar seguimiento. El estudio genético confirma la presencia de la mutación STK11.
DISCUSIÓN
El SPJ es un síndrome autosómico dominante caracterizado por el desarrollo de pólipos gastrointestinales, hiperpigmentación cutánea y predisposición al cáncer en diversos órganos.
La incidencia estimada es de 1 de cada 200 000 nacidos vivos. El gen implicado es el STK11 (19p13.3). Más del 90% de las familias afectadas presentan mutaciones en este gen supresor tumoral2. No existe una clara correlación entre el genotipo y fenotipo.
Las lesiones mucocutáneas pigmentadas aparecen en el 95% de los pacientes como la primera manifestación3. Las más típicas se localizan en la mucosa bucal, aunque también pueden aparecer en otras localizaciones como ojos, narinas o dedos de manos y pies. En la adolescencia algunas de ellas pueden desaparecer, salvo las de la mucosa oral, que suelen persistir a lo largo de toda la vida. Estas lesiones suelen estar presentes desde los dos años de vida y existe un intervalo de tiempo de unos diez años hasta la aparición de pólipos gastrointestinales3.
Los pólipos gastrointestinales aparecen en el intestino delgado (50%), estómago (36%) y colon (21%). Estos pólipos también pueden aparecer fuera del tracto digestivo, como en la vesícula, la vejiga, los bronquios o los uréteres. Los pólipos son benignos, pero pueden causar sangrado, anemia, prolapso rectal o invaginación intestinal.
El diagnóstico clínico se realiza si existen: (1) dos o más pólipos con anatomía patológica típica de SPJ (hamartomatosos); (2) cualquier número de pólipos con antecedentes familiares de SPJ; (3) alteraciones mucocutáneas típicas con antecedentes familiares de SPJ; o (4) cualquier número de pólipos asociado a las alteraciones mucocutáneas típicas.
El diagnóstico se confirmará con la presencia de la mutación en el gen STK11 (19p 13.31).
Debido a la herencia autosómica dominante de esta patología, el paciente deberá acudir a genética para el asesoramiento genético y valorar estudio de familiares.
Estos pacientes precisan un seguimiento muy específico durante toda la vida. Según las recomendaciones de la ESPGHAN4, el control con endoscopia y colonoscopia debe comenzar no más tarde de los 8 años en niños asintomáticos desde el punto de vista gastrointestinal, y antes si presentan síntomas. Estas pruebas se deben repetir cada 3 años, aunque debe ser individualizado.
En la edad pediátrica, el principal riesgo es el desarrollo de una invaginación intestinal cuyo riesgo acumulado es del 50 al 68% durante la infancia, con una edad media entre 10 y 16 años.
Los pólipos mayores de 1,5 cm están asociados a un mayor riesgo de complicaciones, como la invaginación, por lo que las últimas recomendaciones indican que deber ser extirpados, eligiendo de forma individualizada la técnica más apropiada5.
Es muy importante que tanto la familia como su pediatra sospechen una invaginación intestinal cuando el paciente presente clínica compatible, ya que deberá ser remitido urgentemente para realizar una reducción quirúrgica. La reducción mediante enema o aire están contraindicadas.
El desarrollo de cáncer en la edad pediátrica es excepcional, por lo que, en este sentido, no precisan realizar pruebas específicas. Se han descrito tumores de ovario en mujeres y de células de Sertoli en varones, por lo que se debe hacer un especial seguimiento de la pubertad y valorar en varones posibles signos de feminización que nos pongan en alerta.
En la edad adulta, el riesgo de cáncer es muy elevado6 y se pueden desarrollar varios tipos de cáncer (colorrectal, de mama, de páncreas…), por lo que el seguimiento estrecho y diagnóstico precoz serán determinantes para aumentar la supervivencia7.
Como consideraciones finales hay que destacar la importancia de conocer esta patología y sospechar su presencia ante pacientes con máculas hiperpigmentadas orales, similares a las que se muestran en este caso y especialmente en aquellos que ya han presentado sintomatología gastrointestinal compatible. Debemos recordar que en Pediatría se debe hacer una exploración física completa de los pacientes, ya que en muchas ocasiones el diagnóstico nos lo aportan signos o síntomas que no son inicialmente el motivo de consulta. En el SPJ una sospecha clínica precoz conduce a un diagnóstico temprano y a un manejo correcto, que disminuye drásticamente la tasa de complicaciones, como la invaginación intestinal y el desarrollo de cáncer en estadios avanzados.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Contribución de los autores: asistencia clínica, revisión bibliográfica y redacción del artículo (PFS), asistencia clínica y redacción del artículo (DCC), redacción del artículo (CNC, MLJ), revisión bibliográfica (ICF).
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
SPJ: síndrome de Peutz-Jeghers.
BIBLIOGRAFÍA
- Rodríguez Lagos FA, Sorlí Guerola JV, Romero Martínez IM, Codoñer Franch P. Register and clinical follow-up of patients with Peutz-Jeghers syndrome in Valencia. Rev Gastroenterol Mex (Engl Ed). 2020;85(2):123-39.
- Aretz S, Stienen D, Uhlhaas S, Loff S, Back W, Pagenstecher C, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26(6):513-9.
- Xu ZX, Jiang LX, Chen YR, Zhang YH, Zhang Z, Yu PF, et al. Clinical features, diagnosis, and treatment of Peutz-Jeghers syndrome: Experience with 566 Chinese cases. World J Gastroenterol. 2023;29(10):1627-37.
- Latchford A, Cohen S, Auth M, Scaillon M, Viala J, Daniels R, et al. Management of Peutz-Jeghers Syndrome in Children and Adolescents: A Position Paper From the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019;68(3):442-52.
- Latchford AR, Neale K, Phillips RK, Clark SK. Peutz-Jeghers syndrome: intriguing suggestion of gastrointestinal cancer prevention from surveillance. Dis Colon Rectum. 2011;54(12):1547-51.
- Van Lier MG, Wagner A, Mathus Vliegen EM, Kuipers EJ, Steyerberg EW, Van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258-64.
- Jelsig AM, van Overeem Hansen T, Gede LB, Qvist N, Christensen LL Lautrup CK, et al. Survival, surveillance, and genetics in patients with Peutz-Jeghers syndrome: A nationwide study. Clin Genet. 2023;104(1):81-9.