
Vol. 24 - Num. 95
Casos clínicos en Digestivo
Ictericia en el lactante: más allá de la atresia de vías biliares
Irene Gómez-Pastrana Paua, Itziar Bueno Vidána, Enrique Medina Beníteza, Sonia Mayo de Andrésb, Enrique Salcedo Lobatoa, Iván Carabaño Aguadoa
aSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
bServicio de Genética Clínica. Hospital Universitario 12 de Octubre. Madrid. España.
Cómo citar este artículo: Gómez-Pastrana Pau I, Bueno Vidán I, Medina Benítez E, Mayo de Andrés S, Salcedo Lobato E, Carabaño Aguado I. Ictericia en el lactante: más allá de la atresia de vías biliares . Rev Pediatr Aten Primaria. 2022;24:311-5.
Publicado en Internet: 04-10-2022 - Número de visitas: 14855
Resumen
La colestasis es una entidad que se define por la elevación de bilirrubina conjugada sérica (más de 2 mg/dl o más del 20% del total) y que suele acompañarse de clínica de ictericia, coluria y acolia/hipocolia. Existen múltiples entidades causantes de tal cuadro, pero en el caso de la colestasis neonatal, es fundamental descartar con urgencia la atresia de vías biliares extrahepáticas (AVBE), ya que precisa una intervención quirúrgica de derivación de flujo biliar de forma temprana por sus implicaciones pronósticas futuras. Ante una colestasis neonatal en la que se ha descartado la AVBE, el diagnóstico diferencial se realizará en función de la evaluación de distintos factores, tales como la cifra de gamma-glutamil transferasa (GGT), el valor de los ácidos biliares, si hay o no sospecha de enfermedad metabólica o por la presencia de otras anomalías asociadas.
Palabras clave
● Atresia vías biliares ● Colestasis ● IctericiaINTRODUCCIÓN
Se presenta a continuación el caso clínico de una lactante de 1 mes y medio que, entre la tercera y cuarta semana de vida, comienza con un cuadro ictericia progresiva, objetivándose parámetros analíticos compatibles con colestasis con elevación de bilirrubina directa (BD) y ácidos biliares, con GGT dentro de la normalidad y coagulopatía. Se expone a continuación el manejo clínico de la paciente, así como los distintos diagnósticos diferenciales y el diagnóstico final.
CASO CLÍNICO
Lactante de 1 mes y 17 días, a término y sin antecedentes médico-quirúrgicos de interés, con pruebas metabólicas dentro de normalidad, remitida a Urgencias desde el centro de salud, por ictericia en progresión, desde los 25 días de vida. Los días previos a la consulta comienza con distensión abdominal leve. Ausencia de coluria o acolia, fiebre, vómitos o alguna otra clínica infecciosa. No presenta irritabilidad ni rechazo de tomas. Alimentada con lactancia materna exclusiva, con adecuada ganancia ponderal. La madre refiere que no ha habido consumo de productos de herbolario, alcohol, tabaco u otros tóxicos.
A la exploración inicial destaca ictericia conjuntival y cutánea hasta abdomen, hepatomegalia a 1-2 cm del reborde costal derecho y palpación de polo de bazo. Ante ictericia con hepatomegalia en lactante mayor de 15 días de vida se extrae analítica con hemograma, bioquímica y coagulación. En analítica presenta hemograma sin alteraciones (Hb 10,9; plaquetas 564 000; leucocitos 12 000), en bioquímica se objetiva bilirrubina total (BT) de 7 mg/dl con bilirrubina directa (BD) de 5,6 mg/dl, elevación de transaminasas GPT 413 U/l y GOT 711U/l con GGT dentro de la normalidad (31 U/l) y FA 717 U/l. Coagulación alterada, con actividad de protrombina de 12,7% e INR 5,2.
Ante una colestasis con coagulopatía en el lactante, se administra una dosis de vitamina K intravenosa; y como estudio etiológico inicial, se realiza una ecografía abdominal que confirma hepatomegalia de 8 cm con edema periportal y esplenomegalia de 7 cm, con visualización de vía biliar intra y extrahepática sin dilatación y sin signos indirectos de atresia de vías biliares.
La paciente ingresa en planta de hospitalización para completar estudio etiológico de cuadro colestático. Tras una dosis única de vitamina K corrige inicialmente la coagulopatía. Se completa estudio con nueva determinación analítica, en la que persiste elevación de BT a expensas de BD, elevación de transaminasas, aunque en descenso, normalización de coagulación y elevación de ácidos biliares hasta 166,86 µmol/l. Se realiza despistaje de enfermedades causantes de cuadro de colestasis, con determinación de alfa-1-antitripsina (en rango de normalidad) y virus hepatotropos e infecciones connatales (TORCH) negativas. Se extrae panel genético para estudio de colestasis intrahepáticas familiares. Se inicia tratamiento con vitaminas liposolubles y con ácido ursodesoxicólico a 15 mg/kg/día y se indica biopsia hepática. En la misma se observa una intensa hepatitis colestásica con transformación gigantocelular y leve ductopenia.
Durante el ingreso, la paciente permanece con buen estado general y sin afectación neurológica, por lo que se traslada a domicilio con seguimiento estrecho en consultas externas. Se constata empeoramiento de la ictericia, prurito con irritabilidad y progresión de hepatomegalia (4-5 cm de reborde costal derecho) y esplenomegalia (2-3 cm del reborde costal izquierdo). Finalmente, los resultados genéticos son compatibles con colestasis intrahepática familiar tipo 2 (PFIC 2), con mutación genética en homocigosis en el gen ABCB11.
La paciente se encuentra en una fase estable, aunque progresiva de la enfermedad, con controles frecuentes y en tratamiento con soporte nutricional, vitaminas liposolubles, ácido ursodesoxicólico y rifampicina.
DISCUSIÓN
Fisiológicamente, la colestasis se define como un proceso en el que existe alteración del flujo biliar, evidencia histológica de depósito de pigmentos biliares en los hepatocitos y conductos biliares y aumento de concentración sérica de los productos excretados por la bilis. La colestasis se caracteriza por una alteración de la función hepática con elevación de la bilirrubina directa sérica >2 mg/dl o >20% de la bilirrubina total en caso de lactantes pequeños. Clínicamente, asocia una ictericia de progresión céfalo-caudal, así como distintos grados de acolia/hipocolia y coluria y que, en función de la enfermedad subyacente que ocasione el cuadro, puede asociar otra sintomatología acompañante como prurito -característico de las colestasis intrahepáticas familiares progresivas (PFIC)- o xantomas1.
Ante un neonato mayor de 2 semanas de edad con ictericia, en especial si no está alimentado con lactancia materna, siempre debe descartarse la existencia de una colestasis: en primer lugar, mediante un análisis de sangre con perfil hepático y bilirrubina directa. Una vez confirmada esta, es primordial confirmar o descartar con urgencia una atresia de vías biliares extrahepáticas (AVBE). Recordemos la importancia pronóstica de la realización de una técnica de derivación de flujo biliar -portoenterostomía de Kasai- en las 6 primeras semanas de vida, con el fin de evitar el trasplante hepático2,3.
Una vez confirmada la colestasis, deben realizarse de forma escalonada una serie de pruebas diagnósticas más específicas destinadas a esclarecer la etiología y poder realizar así un correcto diagnóstico diferencial. En primer lugar, debería realizarse un análisis que incluya ácidos biliares, hemograma, bioquímica y coagulación, así como una ecografía de la vía biliar de cara a encontrar signos directos o indirectos de una AVBE. Esta entidad se acompaña de elevación de GGT >300 (este hecho confiere un alto valor predictivo positivo)3. En caso de dudas diagnósticas, o si no se ha podido descartar la AVBE, debe realizarse de forma precoz una biopsia hepática percutánea, ya que la colangiografía retrógrada endoscópica es difícil de realizar a estas edades tan tempranas por el tamaño de los canalículos biliares. Si tras esto siguen persistiendo dudas sobre la posibilidad de tratarse de una AVBE y persiste colestasis con elevación de GGT y acolia en un lactante de más de dos semanas, debe realizarse una colangiografía intraoperatoria con visualización directa de la vía biliar1,2.
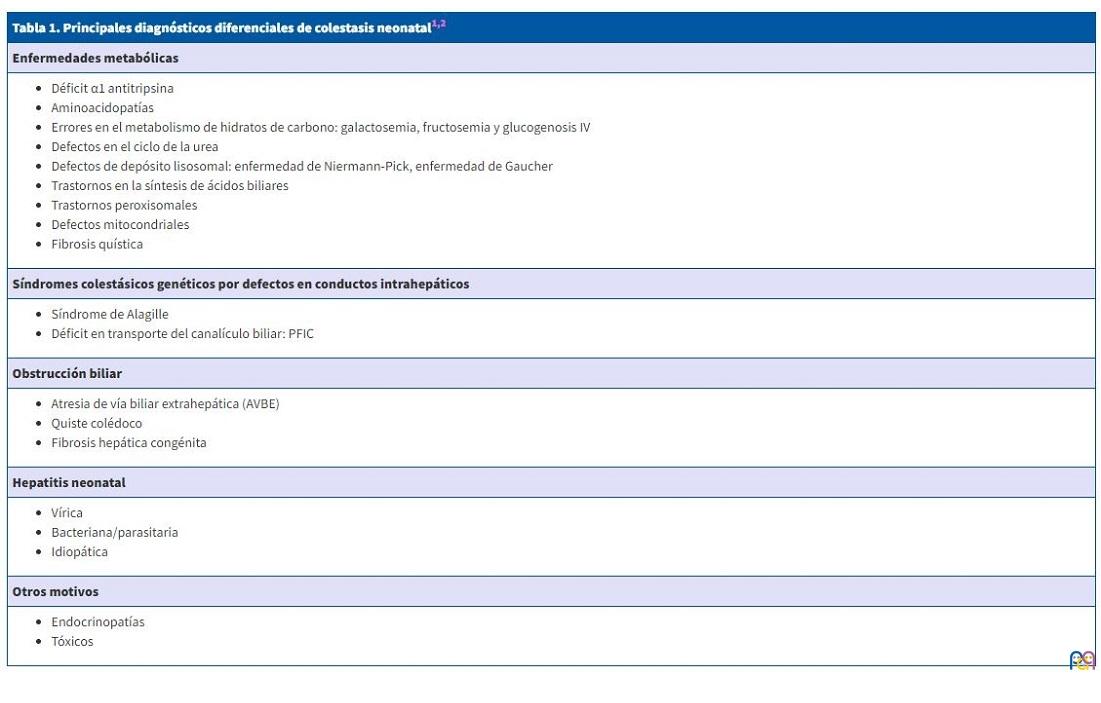
Una vez descartada la AVBE debe realizarse un correcto diagnóstico diferencial de todas las posibles causas de colestasis neonatal o del lactante, exponiéndose a continuación las principales etiologías (Tabla 1).
| Tabla 1. Principales diagnósticos diferenciales de colestasis neonatal1,2 |
|---|
| Enfermedades metabólicas |
|
| Síndromes colestásicos genéticos por defectos en conductos intrahepáticos |
|
| Obstrucción biliar |
|
| Hepatitis neonatal |
|
| Otros motivos |
|
El manejo de estos pacientes dependerá de la enfermedad de base que ocasione la colestasis, pero independientemente de ello, siempre que se trate de una causa de colestasis intrahepática y, por tanto, no abordable por cirugía, se debe iniciar tratamiento con ácido ursodesoxicólico para favorecer el flujo biliar, así como con vitaminas liposolubles (A, D, E y K), por no absorción de estas; y llevar un control nutricional estricto, valorando individualmente la necesidad de suplementar con triglicéridos de cadena media (MCT). En función de la sospecha diagnóstica, deberán añadirse otros fármacos, así como resincolestiramina o rifampicina, en caso de prurito intenso3,4.
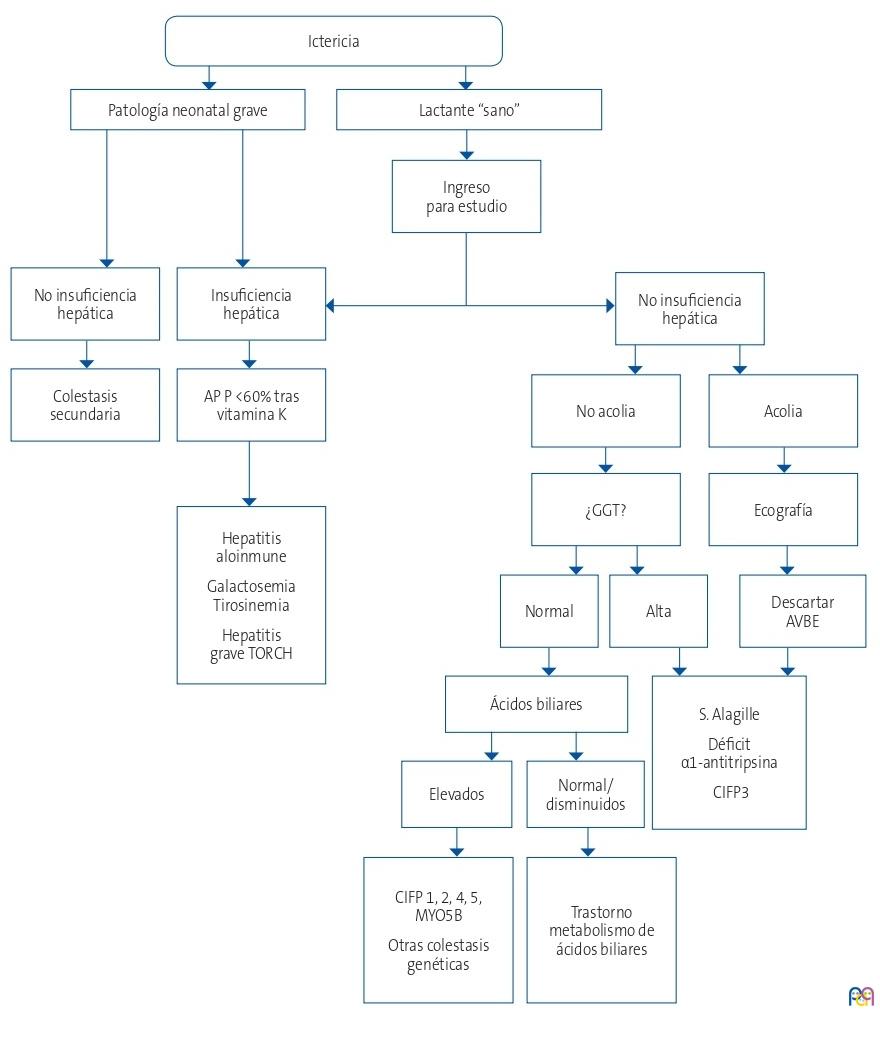
A continuación, se expone un algoritmo diagnóstico de las principales causas de colestasis neonatal en nuestro medio (Fig. 1)4.
| Figura 1. Algoritmo del diagnóstico de colestasis neonatal1,4 |
|---|
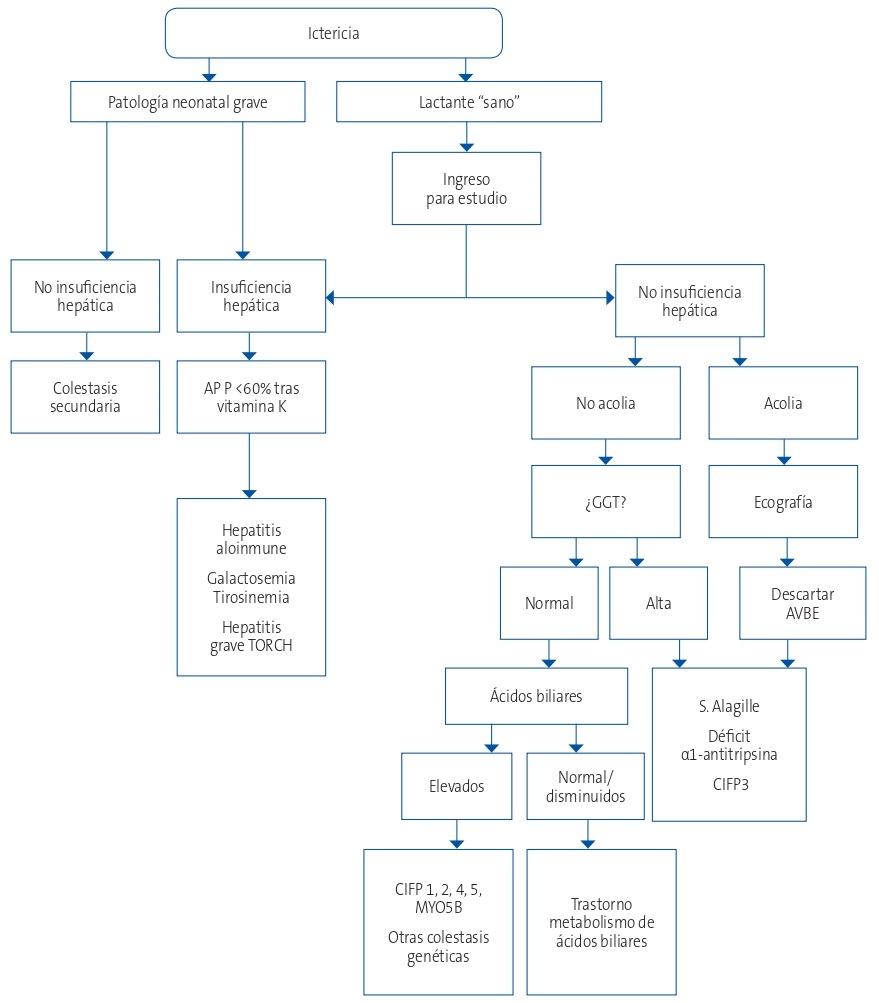 |
Por último, la PFIC es una entidad rara debida a mutaciones genéticas de los sistemas de transporte de canalículos biliares de los hepatocitos causante de síndromes de colestasis crónica a edades muy tempranas con expresiones clínicas distintas en función del gen afecto, pero en las que habitualmente se produce una progresión a cirrosis e insuficiencia hepática en la primera década de la vida. El diagnóstico de esta entidad se realiza mediante secuenciación genética del gen afecto. En el caso de la PFIC2, el gen afectado más común es el ABCB11, localizado en el cromosoma 2 y cuya mutación ocasiona un defecto en la bomba exportadora de sales biliares (BSEP). Este hecho provoca secundariamente un aumento de sales biliares intrahepatocitario. Algunas de las PFIC pueden asociar manifestaciones extrahepáticas, como la diarrea característica de la PFIC1. El tratamiento de todas ellas, más allá del tratamiento médico de la colestasis y de síntomas asociados como prurito, en caso de progresión a cirrosis e insuficiencia hepática, es el trasplante hepático, el cual se suele realizar en las primeras décadas de la vida2,4,5.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
AVBE: atresia vía biliar extrahepática · BD: bilirrubina directa · BSEP: bomba exportadora de sales biliares · BT: bilirrubina total · GGT: gamma-glutamil transferasa · PFIC: colestasis intrahepática familiar progresiva.
BIBLIOGRAFÍA
- Medina E, Manzanares J. Colestasis en el recién nacido y lactante. Orientación diagnóstica. An Pediatr (Barc). 2003;58:162-7.
- Ranucci G, Corte CD, Alberti D, Bondioni MP, Boroni G, Calvo PL, et al. Diagnostic approach to neonatal and infantile cholestasis: A position paper by the SIGENP liver disease working group. Dig Liver Dis. 2022;54:40-53.
- De la Vega A, Frauca Remacha E, Síndrome colestásico. Actitud diagnóstico-terapéutica. Pediatr Integral. 2015;19:168-79.
- Fernández Tomé E, Frauca Remacha E. Colestasis del lactante. Sociedad Española de Gastroenterología y Nutrición. Tratamiento en Gastroenterología, hepatología y Nutrición. 5.ª Edición. Madrid: Ergón; 2021. p. 555-73.
- Hierro l, Jara P. Childhood cholestasis and bile transporters. Gastroenterol Hepatol. 2005;28:388-95.