
Vol. 24 - Num. 94
Originales
Impacto de la drepanocitosis en las familias de los afectados. Estudio cualitativo sobre sus experiencias, percepciones y necesidades
Ana Bernat Nogueraa, Ainhoa Iscar Ortsb, Ana Mascaró Garcíac, Xènia Chela Álvarezd, Trinidad Planas Juane
aPediatra. CS Son Gotleu. Palma . Islas Baleares. España.
bEnfermera. CS Son Gotleu. Palma. Islas Baleares. España.
cMediadora cultural. CS Son Gotleu y Escuela Graduada. Palma. Islas Balerares. España.
dSocióloga. Unidad de Investigación de Atención Primaria de Mallorca. Palma. Islas Baleares. España.
eEnfermera. CS Son Gotleu. Palma de Mallorca. España.
Correspondencia: A Bernat. Correo electrónico: ana.bernat@ibsalut.es
Cómo citar este artículo: Bernat Noguera A, Iscar Orts A, Mascaró García A, Chela Álvarez X, Planas Juan T. Impacto de la drepanocitosis en las familias de los afectados. Estudio cualitativo sobre sus experiencias, percepciones y necesidades. Rev Pediatr Aten Primaria. 2022;24:e207-e215.
Publicado en Internet: 20-06-2022 - Número de visitas: 14617
Resumen
Introducción: la drepanocitosis o enfermedad de células falciformes es una enfermedad genética que afecta mayoritariamente a población de origen africano. Requiere ingresos largos y repetidos, afectando a la calidad de vida. Si la situación socioeconómica es vulnerable, el efecto de la enfermedad en su vida se agrava. Este estudio pretende conocer las preocupaciones, creencias y necesidades de las familias de los menores afectados de drepanocitosis, los conocimientos previos sobre la enfermedad, las repercusiones psicosociales, la satisfacción con la asistencia sanitaria y el posible estigma que rodea esta enfermedad.
Material y métodos: estudio cualitativo mediante entrevistas a familiares de los afectados.
Resultados: se realizaron 20 entrevistas. Se exploró la relación de las familias con la enfermedad, el entorno sociofamiliar, las preocupaciones, emociones y estrategias de afrontamiento, los recursos deseados y no deseados y la satisfacción con la atención recibida. Al analizarlas, destacó el impacto del diagnóstico, un buen conocimiento de los síntomas y tratamiento, y diferencias en el conocimiento de las causas de la enfermedad. El rol de cuidador está ligado al género femenino. La organización familiar y laboral es causa frecuente de estrés. Se detectan preocupaciones por la enfermedad y el día a día, emociones y sentimientos negativos y positivos, así como diferentes estrategias de afrontamiento. Expresan recursos deseados como ayudas por hijo/a enfermo/a. Valoran positivamente la atención sanitaria recibida.
Conclusiones: las entrevistas en profundidad con las familias de las personas afectadas ayudan a conocer mejor su realidad y sus necesidades, mejorando así la atención a los menores con drepanocitosis.
Palabras clave
● Calidad de vida ● DrepanocitosisINTRODUCCIÓN
La drepanocitosis o enfermedad de células falciformes (ECF) es una enfermedad crónica que cursa con complicaciones agudas. Es una hemoglobinopatía hereditaria caracterizada por la presencia de hemoglobina falciforme (HbS). Se transmite de forma autosómica recesiva. Afecta mayoritariamente a personas de origen africano1,2. Debido a los movimientos poblacionales actuales, es ahora una enfermedad global3,4. Se estima que cada año nacen unos 300 000 niños y niñas con ECF3, siendo la alteración genética más prevalente en los cribados neonatales en varios países2. En determinadas áreas de África, un 50% de estos pacientes muere antes de los 5 años, sin tener datos de la supervivencia global5.
Las complicaciones derivan de la hemólisis y/o vasooclusión, causando anemia, asplenia funcional, accidentes cerebrovasculares y crisis dolorosas. Estas complicaciones resultan de infecciones, deshidratación, cambios de temperatura, estrés o no tienen desencadenante claro2,3. Los afectados precisan ingresos múltiples, frecuentemente prolongados, tratamientos crónicos y visitas hospitalarias continuadas. El diagnóstico precoz6, las vacunaciones, el tratamiento antibiótico profiláctico, las transfusiones crónicas, la hidroxiurea, el trasplante de células hematopoyéticas y la atención multidisciplinar han disminuido de manera importante la morbimortalidad2 y han contribuido a un aumento de su esperanza de vida. Nuevos tratamientos, como crizanlizumab y voxelotor, podrían también hacerlo.
La ECF tiene implicaciones personales, familiares, sociales y laborales2-4,7-12, y disminuye la calidad de vida del paciente y su familia 2,13,14. Muchas sufren aislamiento social, llevando la enfermedad en secreto por la creencia de que han sido castigadas3,8,11. Varios estudios refieren que una atención integral con comunicación entre niveles asistenciales podría mejorar la calidad de vida de los niños y sus familias3,10,11,15, así como un mayor conocimiento de la enfermedad3,6, ayuda psicológica y contacto con otras familias afectadas3,8,11. Se puede afirmar que mejorar el apoyo a las familias constituye una parte importante del tratamiento14.
Al tratarse de una enfermedad más prevalente en una población vulnerable en el momento sociodemográfico actual, se considera que es tributaria de una atención especial para poder garantizar la equidad.
Este estudio pretende conocer las preocupaciones, creencias y necesidades de las familias de los menores atendidos, los conocimientos que tienen sobre la enfermedad, sus problemas psicosociales y sus vivencias en torno a la enfermedad, así como los estigmas y mitos que la rodean y su relación con el sistema sanitario.
MATERIAL Y MÉTODOS
Se realizó un estudio cualitativo con enfoque fenomenológico mediante entrevistas individuales semiestructuradas a familiares de pacientes con ECF menores de 14 años diagnosticados antes de 2016.
Los participantes se seleccionaron de las bases de datos de pacientes del Hospital Son Espases y Hospital Son Llàtzer, los únicos centros especializados donde se hace su seguimiento en Baleares. El equipo investigador contactó telefónicamente (dos intentos a diferentes horas) con un familiar, explicándole el estudio e invitándole a participar. Con aquellos que aceptaron, se concertó cita para realizar la entrevista, adaptándose en lugar y horario a sus preferencias. Se entregó una hoja informativa y se firmó el consentimiento informado. Al finalizarla, se ofreció un obsequio por familia como agradecimiento. El guión de la entrevista se basó en los objetivos del estudio y en la revisión bibliográfica. Las dimensiones exploradas fueron: conocimiento de la enfermedad, afrontamiento, valoración de la atención y valoración del presente y futuro. Se recogieron también variables sociodemográficas (relación con el menor, país de origen, etnia, etc.).
Las entrevistas se realizaron entre septiembre de 2017 y agosto de 2018, duraron entre 15 y 60 minutos, fueron grabadas en audio y transcritas literalmente, asignándoles un código y guardándolas en archivos encriptados sin datos personales para garantizar el anonimato. La mayoría de las entrevistas fueron en castellano. Algunas se realizaron en inglés por parte de la mediadora cultural. Todo el proceso (realización de entrevistas, transcripción y traducción) se llevó a cabo por el equipo investigador.
Se realizó un análisis de contenido elaborando un árbol de códigos a partir de los objetivos y la lectura de las entrevistas. El análisis lo realizaron tres investigadores por separado. Posteriormente, se pusieron en común los resultados consensuándose las diferencias, garantizando la validez interna. Se utilizó el programa informático Nvivo.
Este estudio fue aprobado por el Comité de Ética de Investigación de las Islas Baleares (n.º IB-3428/17).
RESULTADOS
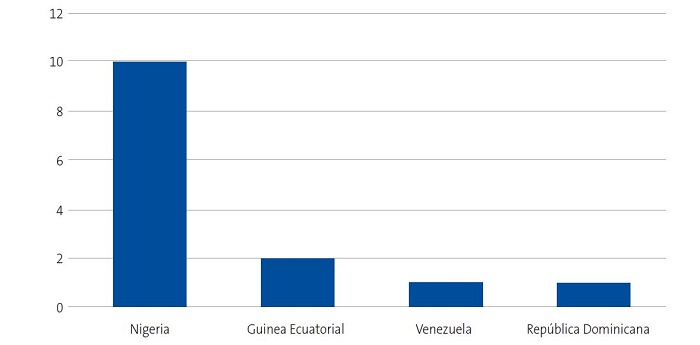
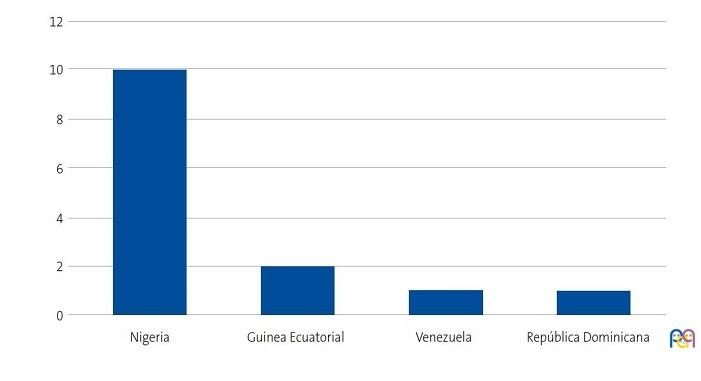
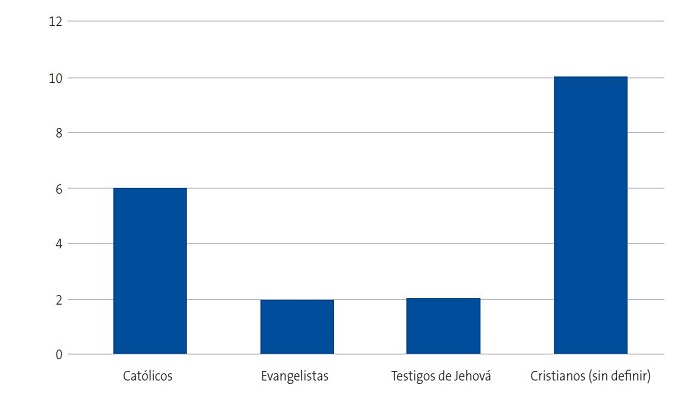
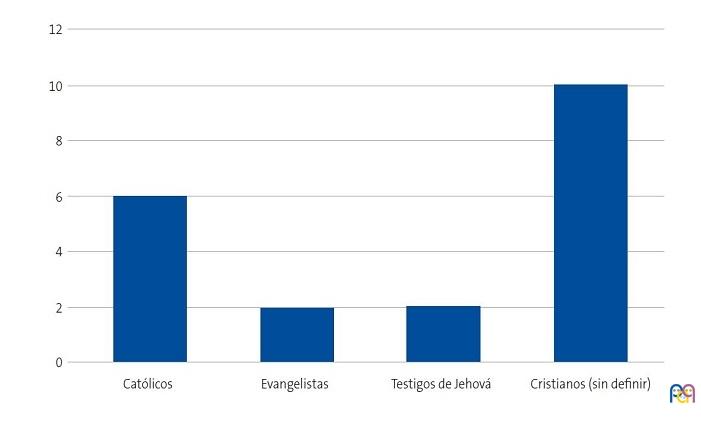
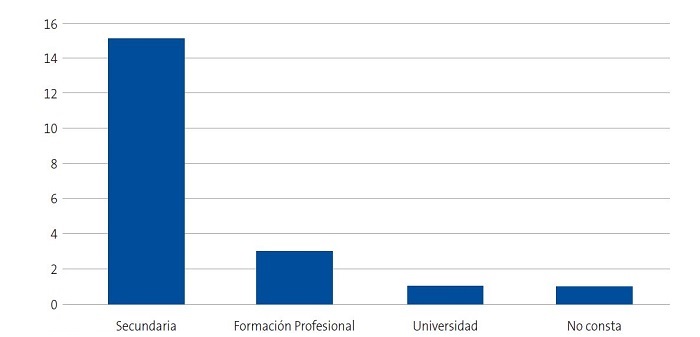
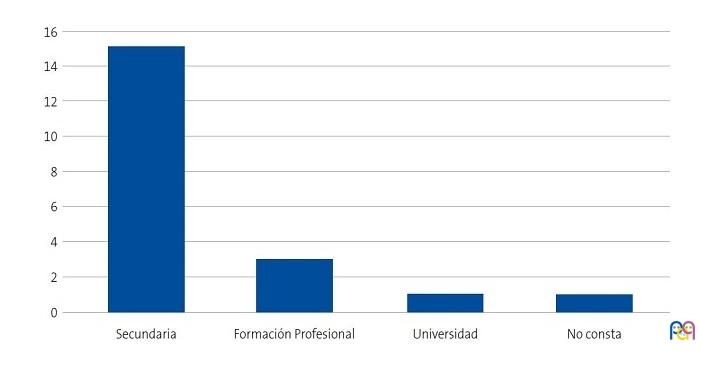
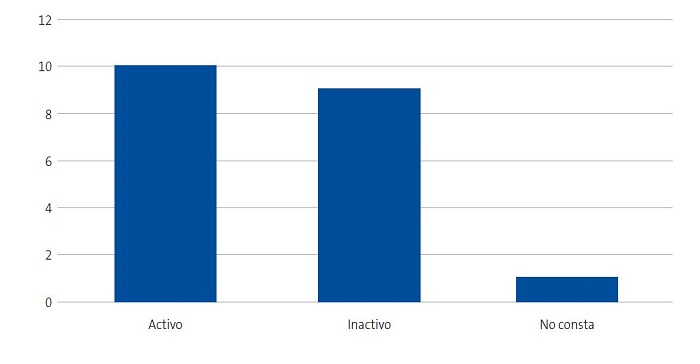
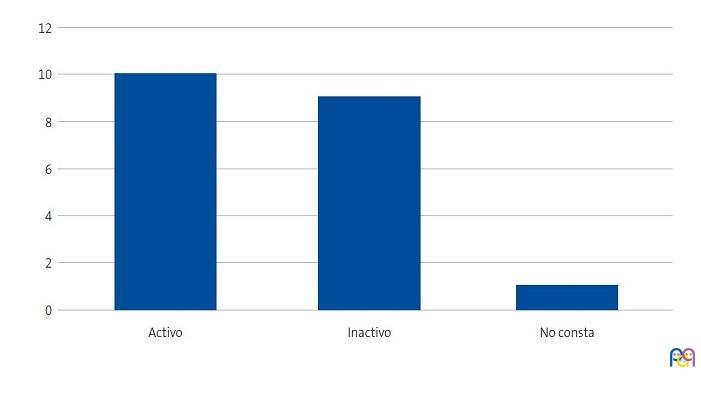
Se detectaron 28 pacientes menores de 14 años con diagnóstico de ECF. Se consiguió contactar con las familias de 15 menores (dos de ellos hermanos), de los cuales padre, madre y/u otro familiar accedieron a entrevistarse. Se realizaron 20 entrevistas a un total de 14 familias, participando 11 madres, 4 padres y 5 familiares convivientes. Se pudo entrevistar a ambos padres en tres ocasiones, solo a la madre en ocho ocasiones, y en una, solo al padre. Sus características sociodemográficas se presentan en las Figs. 1, 2, 3 y 4.
| Figura 1. Estudio sobre las preocupaciones, creencias y necesidades de las familias de niños con anemia de células falciformes (drepanocitosis). Origen de las familias entrevistadas |
|---|
 |
| Figura 2. Estudio sobre las preocupaciones, creencias y necesidades de las familias de niños con anemia de células falciformes (drepanocitosis). Religión de los entrevistados |
|---|
 |
| Figura 3. Estudio sobre las preocupaciones, creencias y necesidades de las familias de niños con anemia de células falciformes (drepanocitosis). Nivel de estudios de los entrevistados |
|---|
 |
| Figura 4. Estudio sobre las preocupaciones, creencias y necesidades de las familias de niños con anemia de células falciformes (drepanocitosis). Actividad laboral de los entrevistados |
|---|
 |
Se exponen los resultados en cuatro áreas: (1) relación con la enfermedad, (2) entorno sociofamiliar y organización del cuidado, (3) preocupaciones, emociones y estrategias de afrontamiento y (4) recursos y satisfacción con la atención recibida.
Relación con la enfermedad
Todos los menores, excepto uno, fueron diagnosticados en España. En general, el momento del diagnóstico fue un momento impactante, expresando miedo, incredulidad y sorpresa, así como falta de comprensión de la situación y de las explicaciones médicas. Algunos lloran cuando lo recuerdan.
"Lo pasamos muy mal porque los primeros momentos tú te sientes como si no estuvieses en tu vida" (E1).
"Devastador, me sentí morir, no hay razón en mí para vivir" (E2).
Casi todos los entrevistados tenían conocimientos generales sobre la ECF (nombre, síntomas, relación con la sangre, etc.). La mayoría conocía el nombre. Previamente al diagnóstico, muchos no la conocían. Aunque solo una minoría refirió que es genética, todos los entrevistados excepto uno se realizaron el estudio para descartar la enfermedad o el estado de portador. Alguno de los entrevistados refirió explícitamente que la enfermedad no tiene cura, aunque se detectó en otras entrevistas dificultades para entender el concepto de cronicidad.
"Yo entiendo en mis estudios que cuando alguien AS no es bueno tener hijos con alguien que también es AS porque luego puedes tener un hijo SS que es sickle cell disease" (E1).
"No es una enfermedad que se puede decir contagiosa" (E10).
"Es una enfermedad que primero no tiene cura" (E10).
Los que no la conocían, después del diagnóstico de su familiar, supieron que en su país había casos. Tres familias declararon que hay más casos familiares de drepanocitosis y explican que la información recibida aquí difiere de la de su país.
"La anemia que ellos conocen es diferente, lo que ellos más conocen así es lo grave, que viene de repente" (E14).
"Esta enfermedad es frecuente en África, no he visto en blancos" (E2).
Prácticamente todos supieron identificar los síntomas de alarma y la importancia del tratamiento.
"El ojo puede tener amarillo y el cuerpo cuando está muy flojo o cuando me dice «mamá dolor aquí», corriendo a hospital, o cuando tiene fiebre muy alta, corriendo al hospital" (E1).
"Tiene que tomar mucha agua, no puede hacer ejercicio muy, muy, muy forzoso" (E8).
Entorno de la familia y organización de los cuidados
Todos los casos de nigerianos de etnia Edo no han querido explicar a su entorno que su hijo está enfermo y en la mayoría esta información está limitada al núcleo familiar más íntimo (madre y padre). La única madre nigeriana de etnia Urhobo no refirió problemas en explicar la enfermedad de su hija. Las familias de otras etnias declararon informar a toda la familia y al colegio.
"No quiero que los africanos sepan, quiero que no sepan todo lo que concierne a mi hijo. No quiero que la gente de mi país sepa porque nuestra gente se reiría de ti" (E2).
En la mayoría de los casos, la principal cuidadora es la madre o alguna mujer, independientemente de su situación laboral. También es la madre la que se queda con el menor durante los ingresos, siguiendo además con el cuidado de los hermanos y con las tareas domésticas. Esto supone un estrés en la organización familiar. Cuatro mujeres declararon ser las responsables únicas, mientras que nueve dijeron tener algún tipo de ayuda. Una mujer refirió que su marido no solo no le ayuda, sino que pedirle ayuda le causa más estrés y complicaciones.
"Hombre no es como mujer, si hombre estar en casa o mujer estar en casa hay diferencia, porque tú vienes aquí a cuidar por el día o por la noche, luego voy a casa a cocinar, cuidar a los demás, lavar ropa, cocinar para guardar en la nevera y luego tú vienes a casa y esto es lo que no entiende" (E3).
Al ser las madres las principales cuidadoras, son ellas quienes más arriesgan su trabajo para cuidar. Para acudir a las consultas piden permiso en el trabajo o lo hacen coincidir con el día libre. En un caso, la entrevistada está buscando un trabajo con horario compatible con el cuidado de su hijo.
"Cuando tiene fiebre enseguida me vengo. He tenido algunos problemas con la empresa que me dicen «tienes que avisar» y yo «sí, pero primero es mi hijo, no te puedo avisar si le da a media noche»" (E4).
"Si ella tiene cita por ejemplo mañana o en una semana (…) si no es mi día libre yo no puedo o (…) yo hablar con mi jefe si me dan este tiempo de ir a buscarla, ir al médico y volver a trabajar" (E3).
Cuando los hombres ayudan tienen un papel secundario, complementando las actividades de la madre. Solo en un caso el padre declara ser corresponsable de la enferma junto a la madre repartiéndose las tareas de cuidado.
"Mi mujer está allí, yo me voy a casa, pero no sé cocinar" (E5).
"Visitas siempre mi mujer" (E14).
"Podría decir que la madre cuida mejor del niño" (E5).
Las dos familias de Guinea Ecuatorial explicaron que reparten el cuidado del menor con la familia extensa, al no estar sus padres en Mallorca, aunque la responsable principal es una mujer.
Preocupaciones, emociones y estrategias de afrontamiento
Las principales preocupaciones detectadas estuvieron relacionadas con el dolor y sufrimiento del familiar, la posible muerte prematura, la incertidumbre por el futuro y los posibles efectos secundarios del tratamiento. Otras preocupaciones minoritarias fueron la vida académica y la posible afectación en la fertilidad de las niñas. Algunas familias nigerianas (Edo y Urhobo) declararon no sentirse preocupadas porque ven al menor bien, siguiendo los controles médicos correctamente y confiando en el tratamiento y en Dios.
"Con esta enfermedad yo preocupar mucho y con tanto sufrir yo sufrir con eso" (E1).
"A veces yo pienso si va a morir" (E12-13).
"Ahora no estoy preocupada, con el medicamento y todo está bien, muy bienv (E6).
Las emociones referidas fueron mayoritariamente negativas: miedo, preocupación, incertidumbre y tristeza. También expresaron sentimientos positivos como confianza, gratitud, felicidad cuando ven que el niño/a está bien, tranquilidad por la atención sanitaria de calidad y porque ven mejoría con la medicación. Muchos tienen la esperanza de mejoría en el futuro.
"Siempre tengo miedo" (E2).
"Cuando ella está enferma a veces me siento triste" (E5).
"Siempre que me dicen (…) que está un poco mejor estoy contenta" (E2).
A lo largo de las entrevistas se evidenciaron factores que les ayudan a llevar mejor la enfermedad:
- Creencia y esperanza en que Dios les ayudará, aunque en un caso una madre refirió que la enfermedad es un castigo de Dios. "Dios va a ayudar" (E12-13), "Dios puede hacer cualquier cosa, tomando pastillas y eso… el resto es Dios, por eso no preocupar" (E14), "¿Por qué es así Dios con la vida de la gente?" (E9).
- Seguimiento médico: la información, consejos y control médico les tranquiliza. "Me han dado esperanza, cuando está enfermo en casa tengo el sentimiento que cuando llegue al hospital se pondrá mejor" (E2).
- El amor hacia el/la niño/a y el soporte familiar les fortalece. "Ella es mi felicidad" (E9), "Ha sido un niño muy ¿cómo decir? querido y con todo ese amor (…) nos entregamos con todas las fuerzas que tenemos" (E11).
- Negación de la enfermedad. "No creo en enfermedad" (E3).
Recursos y satisfacción con la atención recibida
Los entrevistados están satisfechos con la atención recibida: refieren buen trato y confianza en los profesionales. La mayoría de las familias tienen poco apoyo social y familiar. Solo dos familias refieren apoyo de amistades. Un caso puntual ha recibido ayuda de voluntarios de una ONG durante los ingresos, valorándola positivamente. Tres familias declararon recibir algún tipo de ayuda económica.
"Las personas como ustedes me han ayudado un montón" (E4).
Muchos de los entrevistados agradecerían recibir comida durante los ingresos o poder ir a casa a coger comida. También reclamaron medicación gratuita, ayudas para el transporte al hospital, reconocimiento de la ECF como enfermedad crónica con las ayudas pertinentes, rapidez en los resultados de las pruebas realizadas, disminuir las visitas hospitalarias y la posibilidad de días libres en el trabajo para dedicarse al cuidado. Dos entrevistados desearían ayuda psicológica y más comprensión. Un entrevistado preferiría que le hablaran en inglés para entender mejor las explicaciones. Valoraron positivamente que la atención sea gratuita y que la medicación no sea cara.
"Necesitaríamos ayuda de una persona que a lo mejor pueda responsabilizarse [de mi hijo enfermo] cuando no estoy" (E10).
"Estos niños (…) yo veo que no están legalizados con lo que tienen y es una lucha diaria que la familia tiene" (E11).
Al preguntarles explícitamente si estarían interesados en recibir formación sobre la enfermedad y en crear un grupo de apoyo con otras familias, la mayoría mostró interés. Sin embargo, las familias de etnia Edo no asistirían si participaran otras familias de su misma etnia o nacionalidad aduciendo que los demás hablarían de sus problemas y podrían utilizar la información personal para hacerles daño.
Referido al grupo de apoyo: "[Sí,] para darles coraje… pero si son nigerianos, no" (E9); "Si [otra familia] tiene más experiencia que yo me pueden ayudar, puedo aprender algo de ellos (…) también yo decirles mi experiencia" (E6).
Referido al curso de formación: "Mientras más uno sepa creo que mejor" (E8); "Para preocuparme menos" (E9).
DISCUSIÓN
Este estudio muestra cómo el diagnóstico de ECF representa un acontecimiento traumático con importante ansiedad. Se detecta un buen conocimiento de los síntomas y de la importancia del tratamiento, pero relativa falta de conocimiento de la causa genética y de la cronicidad. Los entrevistados refirieron preocupación por el futuro, dificultades organizativas familiares y laborales (sobre todo en el caso de madres, principales cuidadoras), poco soporte social y, en ocasiones, también familiar. Se detecta cierto aislamiento al llevar la enfermedad como un secreto, más visible en familias nigerianas de etnia Edo. En general, la relación de las familias con el sistema sanitario es satisfactoria, aunque se identifican diferentes necesidades que consideramos se deberían tener en cuenta.
El diagnóstico de ECF como un momento impactante y la exposición a diversos estímulos negativos desde el momento del mismo, con aumento de síntomas ansioso depresivos en la familia y en el afectado ha sido un hecho recogido en otros estudios3,6-10. Los cuidados recaen básicamente en la madre o en un personaje femenino, lo que probablemente amplifica las desigualdades de género, ya que es la mujer la principal afectada en cuanto a su dinámica laboral, carga de cuidados y tareas domésticas, conllevando dificultades para la organización, sobre todo durante las crisis agudas. Así consta en diferentes estudios3,11,13. Esto se agrava al ser personas migradas y encontrarse en una situación socioeconómica más vulnerable.
Puede que el insuficiente conocimiento de la causa genética y cronicidad de la enfermedad esté relacionado con la baja comunicación con la familia de temas de salud, llamando la atención especialmente en las familias de etnia Edo. Estos resultados enlazan con los de otros estudios, donde consta que muchas familias llevan la enfermedad como un secreto, sufren aislamiento social y tienen la creencia de que han sido castigados, acompañándose de sentimientos de culpabilidad2,3,11,12.
La mayor limitación de este estudio ha sido la falta de captación y la infrarrepresentación del género masculino, posiblemente por padre no presente y menor implicación en el cuidado del menor. También la barrera idiomática y cultural, minimizándose por la presencia de una mediadora cultural dentro del equipo investigador. Consideramos que existe cierto sesgo de información cuando preguntamos por la atención recibida, al ser las entrevistadoras las profesionales sanitarias que atienden a sus familiares.
Cada vez existen más evidencias de que la pediatría debe encaminarse al cuidado de la unidad familiar para garantizar la salud global de niños/as. Tras el análisis de las entrevistas, y aunque la investigación cualitativa no pretende extrapolar los resultados a la población, consideramos que en la atención de estos pacientes se deben tener en cuenta aspectos como el poco soporte social y trabajos posiblemente precarios, así como la etnia, costumbres y creencias familiares para entender su visión de la enfermedad y mejorar la comunicación, siendo importante la posibilidad de contar con un profesional de la mediación. Los resultados apuntan a líneas de mejora en la atención, como ofrecer ayuda durante los ingresos y facilitar ayudas económicas (como, por ejemplo, la prestación económica por cuidado de menores afectados por cáncer u otra enfermedad grave del Real Decreto 1148/2011, de 29 de julio) y de conciliación con la vida familiar y laboral. Sería interesante plantear a estas familias realizar un curso de formación para aumentar sus conocimientos y/o crear un grupo de apoyo con otras familias y así aumentar el soporte social, aunque hay que tener en cuenta que esta propuesta surge de los profesionales y no de las familias, considerando de nuevo tener en cuenta sus necesidades dependiendo de su contexto cultural.
Concluimos que la atención multidisciplinar, realizando entrevistas clínicas en profundidad centradas en la familia y su situación, solo puede mejorar la calidad de vida de las personas con ECF.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflicto de intereses en relación con la preparación y publicación de este artículo.
Financiación. Estudio financiado por la Comisión de Investigación de Atención Primaria de Mallorca en la IV Convocatoria interna de ayudas para proyectos de investigación de Atención Primaria de Salud en las Islas Baleares 2016 (PI003/17).
ABREVIATURAS
ECF: drepanocitosis o enfermedad de células falciformes.
BIBLIOGRAFÍA
- Rives Sola S. Enfermedad de células falciformes: papel del pediatra. An Pediatr Contin. 2013;11:123-31.
- Enfermedad de células falciformes: guía de práctica clínica. En: Sociedad Española de Hematología y Oncología pediátricas (SEHOP). 2019 [en línea] [consultado el 10/06/2022]. Disponible en www.sehop.org/wp-content/uploads/2019/03/Gu%C3%ADa-SEHOP-Falciforme-2019.pdf
- Keane B, Defoe l. Supported or stigmatised? The impact of sickle cell disease on families. Community Pract. 2016;89:44-7.
- Garcia Arias MB, Cantalejo López MA, Cela de Julian ME, Bravo CR, Galaron GP, Belendez BC. Enfermedad de células falciformes: registro de la Sociedad Española de Hematología Pediátrica. An Pediatr (Barc) 2006;64:78-84.
- Guía de práctica clínica sobre Enfermedad de Células Falciformes Pediátrica. En: Sociedad Española de Hematología y Oncología pediátricas (SEHOP). 2010. [en línea] [consultado el 10/06/2022]. Disponible en www.sehop.org/wp-content/uploads/2013/05/GU%C3%8DA-FALCIFORME-SEHOP-2010.pdf
- Fernández Águila J, Pérez Cogle A, Fragoso M, Rivero Jiménez R. El diagnóstico temprano de la anemia falciforme: un problema no resuelto en África negra. Rev Cubana Hematol Inmunol Hemoter. 2012;28:195-7.
- Serrano Patten AC, Louro Bernald I, Vargas Flores J, Osorio Caballero I, Chavez Perez Teran M. Situacion de salud en familias de niños con padecimiento de anemia drepanocitica en Cuba. Rev Elect Psicol Iztacala. 2008;11:26-55.
- Nika ER, Mabiala Babela JR, Moyen G, Kambouro J. Vécu psychosocial des mères d'enfants drépanocytaires. Arch Pédiatrie. 2016;23:1135-40.
- Dommergues JP, Gimeno l, Galacteros F. Un pédiatre à l'écoute de jeunes adultes drépanocytaires. Arch pédiatrie 2007;17:1115-8.
- Curado MA, Nakheiro MI, Gomes MC, Videira M, Dias P, Gaspar C, Vaz E. Cuando duele… duele de veras. El niño con Drepanocitosis. Enferm Global. 2006;9:1-15.
- Luboya E, Tshilonda J-CB, Bothale Ekila MB, Aloni MN. Repercussions Psychosociales de la drépanocytose sur les parents d'enfants vivant à Kinshasa, République Démocretique du Congo: une étude qualitative. Pan African Med J 2014;19:5.
- Allen TM, Anderson LM, Rothman JA, Bonner MJ. Executive functioning and health-related quality of life pediatric sickle cell disease. Child Neuropsychol. 2017;29:889-906.
- MartínezTriana R, García Hernández A, Guerra González EM, Machado Almeida T, Reytor Alfonso K. Efecto de la drepanocitosis sobre la calidad de vida. Rev Cubana Hematol Inmunol Hemoter. 2015;31:277-87.
- Sehlo MG, Kamfar HZ. Depression and quality of life in children with sickle cell disease: the effect of social support. BMC Psychiatry 2015;15:78.
- Cervera Bravo A, Cela de Julián ME. Anemia falciforme. Manejo en Atención Primaria. Rev Pediatr Aten Primaria. 2007;9:649-68.