
Vol. 24 - Num. 94
Notas clínicas
Dermatomiositis juvenil: a propósito de un caso
Raquel Plácido Paiasa, Raúl Veroz Gonzálezb, Manuel Portillo Márqueza
aServicio de Pediatría. Hospital de Mérida. Mérida. Badajoz. España.
bServicio de Reumatología. Hospital de Mérida. Merida. Badajoz. España.
Correspondencia: R Plácido. Correo electrónico: raquel.placido@salud-juntaex.es
Cómo citar este artículo: Plácido Paias R, Veroz González R, Portillo Márquez M. Dermatomiositis juvenil: a propósito de un caso. Rev Pediatr Aten Primaria. 2022;24:181-6.
Publicado en Internet: 02-06-2022 - Número de visitas: 18067
Resumen
La dermatomiositis juvenil es el subtipo más frecuente del grupo de las miopatías idiopáticas inflamatorias en la edad pediátrica. Clínicamente se caracteriza por la presencia de debilidad muscular proximal y simétrica con afectación cutánea característica. El diagnóstico clásicamente se ha basado en los criterios de Bohan y Peter (1975). Sin embargo, en 2017, el International Myositis Assessment and Clinical Studies Group (IMACS) ha publicado unos criterios de clasificación de las miopatías idiopáticas inflamatorias donde es posible clasificar a un paciente sin necesidad de biopsia. La patogenia de la enfermedad es compleja y no del todo conocida. Están involucrados el sistema inmune innato y adaptativo, donde juega un papel muy importante el interferón, y donde existen agentes externos como infecciones o factores medioambientales que actuarían como desencadenantes en sujetos genéticamente predispuestos.
El tratamiento ha de ser intensivo desde el inicio, con el objetivo de controlar la enfermedad cutánea y muscular de forma precoz, minimizar complicaciones como la calcinosis, más frecuente en niños que en adultos, y mejorar el pronóstico a largo plazo.
Presentamos el caso de una niña prepúber en cuyo debut de la enfermedad solo presentaba dolor e impotencia funcional en pierna izquierda, con antecedente de traumatismo de baja energía, sin elevación de enzimas musculares, lesiones cutáneas ni una biopsia muscular típica.
Palabras clave
● Dermatomiositis infantil o juvenil ● Pápulas de GottronINTRODUCCIÓN
La dermatomiositis juvenil (DMJ) es una miopatía inflamatoria asociada a lesiones cutáneas características1-8. Es una enfermedad sistémica, de naturaleza autoinmune que se inicia antes de los 16 años5. A pesar de su baja prevalencia, representa el 85% de todas las miopatías inflamatorias idiopáticas en niños, con incidencia entre 2-4/1 millón de niños2,7.
La patogenia de esta enfermedad está poco clara1-7. Bohan and Peter (1975) sugirieron 5 subtipos de miositis: dermatomiositis, polimiositis, miositis asociada a neoplasia maligna, dermatomiositis/polimiositis infantil y miositis asociadas a otras conectivopatías1. La clínica cutánea puede preceder a la muscular e incluso se han descrito casos de afectación cutánea exclusiva (DM amiopática)1.
Presentamos el caso de una niña con DM que debuta con dolor muscular sin clínica característica de miopatía inflamatoria al inicio y atribuyéndose el cuadro a un traumatismo previo.
CASO CLÍNICO
Niña de 7 años sin antecedentes personales ni familiares de interés y bien vacunada, que presenta desde hace un mes, y coincidiendo con un traumatismo de baja intensidad, dolor e impotencia funcional en la pierna izquierda, molestias en ambos gemelos y en la cara anterior del muslo. Consultó en el servicio de urgencias al inicio del cuadro, descartándose fracturas. Niega fiebre u otra clínica acompañante.
En la exploración no presenta signos inflamatorios articulares, limitación funcional ni dolor a la movilización, salvo en la zona del recto anterior de la pierna izquierda. Presentaba marcha normal, fuerza y tono muscular conservados, y signo de Gowers negativo. No asociaba lesiones cutáneas y el resto de la exploración era normal.
En la analítica presenta unos reactantes de fase aguda elevados (VSG de 120 mm/hora y PCR de 15,3 mg/l). El hemograma, coagulación, perfil renal y tiroideo, iones y las enzimas musculares (CPK, aldolasa, LDH, AST o ALT) eran normales. El factor reumatoide (FR), HLA-B27, anticuerpos antinucleares (ANA) y anticuerpos antisintetasa resultaron negativos. Presentaba anticuerpos vacunales contra virus de la hepatitis B, varicela zóster, rubeola, sarampión y parotiditis.
Las radiografías de caderas y fémures resultaron normales, así como ecografías de caderas. La ecografía de partes blandas visualiza a nivel distal del gemelo izquierdo, compatible con rotura fibrilar. Se amplió el estudio con resonancia magnética (RM) de todo el miembro inferior izquierdo, apreciándose una imagen sugestiva de edema muscular a nivel anterior del muslo sin objetivarse rotura fibrilar.
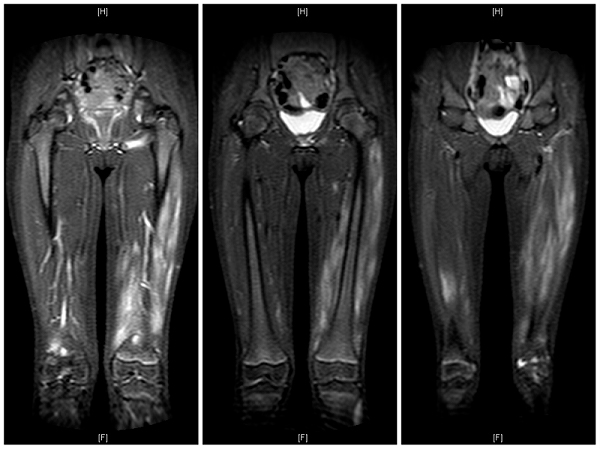
Ante la sospecha de una miopatía inflamatoria, se solicita un electroneurograma y electromiograma (ENG/EMG) de miembros inferiores, ecocardiograma transtorácico, electrocardiograma (ECG) y capilaroscopia, que resultan normales. Se realiza una RM de control que revela datos de edema muscular en la región proximal de ambos miembros inferiores, especialmente en el cuádriceps izquierdo (Fig. 1). La biopsia incisional del músculo cuádriceps femoral derecho demuestra ausencia de actividad inflamatoria, pero con un discreto aumento de lípidos con atrofia leve de fibras tipo 2, sobrexpresión focal de MHC-1 coincidente con una sobrexpresión leve de atrofina y un mínimo componente linfocitario T endomisial, sugiriendo un componente de miopatía inflamatoria en resolución.
| Figura 1. RM de miembros inferiores que revela datos de edema muscular en la región proximal de ambos miembros inferiores, especialmente en el cuádriceps izquierdo |
|---|
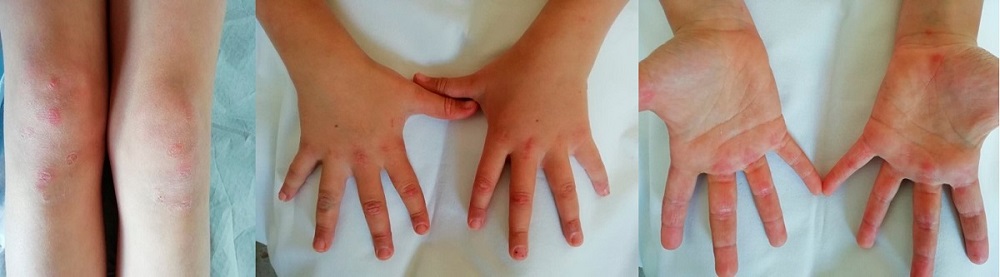 |
Dos meses después, la paciente presenta pápulas de Gottron invertidas en ambas palmas y livedo reticularis en miembros inferiores (Fig. 2).
| Figura 2. Pápulas de Gottron en manos y rodillas |
|---|
 |
Ante la sospecha de DMJ, se inicia metilprednisolona oral (2 mg/kg/día) con mejoría clínica inicial y metotrexato (MTX) a 15 mg semanal subcutáneo con suplementos de folato y calcifediol.
Al año del debut, la paciente se encuentra asintomática en tratamiento con MTX e hidroxicloroquina 50 mg diarios. No obstante, una semana antes a la consulta reaparecen pápulas de Gottron en manos y rodillas (Fig. 2), por lo que se administraron inmunoglobulinas endovenosas con buena respuesta.
Actualmente se encuentra en remisión clínica, libre de esteroides y realiza ejercicio diario con buena tolerancia.
DISCUSIÓN
La debilidad muscular en pediatría presenta un diagnóstico diferencial muy amplio. Se incluyen las distrofias musculares, miopatías metabólicas (glucogenosis, lipidosis, enfermedades mitocondriales), miopatías endocrinológicas o inducidas por fármacos, miastenia gravis, atrofia muscular espinal y otros grupo de enfermedades que, además, pueden o asociar exantemas como infecciones víricas, infecciones bacterianas y parasitarias, enfermedades inflamatorias (celiaquía, enfermedad inflamatoria intestinal) o enfermedades reumáticas inflamatorias (lupus eritematoso sistémico, esclerodermia, artritis idiopática juvenil, enfermedad mixta del tejido conjuntivo, vasculitis o las miopatías idiopáticas inflamatorias crónicas)1.
La dermatomiositis juvenil (DMJ) se caracteriza por debilidad muscular proximal y hallazgos cutáneos distintivos, pero, debido al inicio insidioso de los síntomas, en muchos casos, es posible que no toda la clínica se presente desde el inicio de la enfermedad, siendo difícil el diagnóstico, ya que puede confundirse con un proceso viral5.
La etiología de la DMJ es poco conocida1. Las teorías actuales contemplan una combinación de factores externos (neoplasia maligna, infecciones, fármacos…), disfunción inmune y respuestas tisulares específicas que involucran endotelio de músculo, piel y vasos pequeños en individuos genéticamente susceptibles1,3,5,6. Se ha descrito la asociación a determinados alelos HLA (B8, DRB1*0301, DQA1*0501 y DQA1*0301) y a algunos polimorfismos en el gen del factor de necrosis tumoral alfa (TNF-α)5,6.
La clínica es muy heterogénea. Al comienzo puede existir malestar general, fatiga, anorexia e irritabilidad. La debilidad muscular, principalmente proximal en cintura escapular y pelviana, suele ser de inicio insidioso con una evolución progresiva a lo largo de varios meses. En la exploración pueden apreciarse los signos de Gowers y Trendelemburg5. Entre el 10-25% de los pacientes pueden tener afectación de la musculatura orofaríngea, con disfonía, disfagia y voz nasal, conllevado un alto riesgo de broncoaspiración y/o atragantamiento1,5.
Las manifestaciones cutáneas constituyen la clave para el diagnóstico de la enfermedad, pudiendo aparecer antes o después de la debilidad. El eritema heliotropo (exantema eritematovioláceo localizado en los párpados superiores) y las pápulas de Gottron (pápulas rosadas o violáceas en la superficie extensora de las manos, codos, rodillas y maléolo medial) son patognomónicas y se observan hasta en el 80% de los pacientes5. Algunos pacientes también desarrollan telangiectasias en párpados y eritema malar. Las alteraciones en los capilares periungueales son características y reflejan la presencia de una vasculopatía sistémica1,5.
Otras presentaciones menos frecuentes son el eritema en forma de V en la parte superior del tronco o difuso en tronco y extremidades, la lipodistrofia, la calcinosis de tejidos blandos, asociados a anticuerpos anti-NXP, y relacionada con la gravedad de la enfermedad cutánea y la presencia de vasculopatía1,5.
A nivel sistémico puede haber afectación de órganos muy diversa, principalmente afectación pulmonar intersticial o gastrointestinal en forma de vasculitis1,5.
El diagnóstico es clínico y, clásicamente, se han utilizado los criterios de clasificación propuestos por Bohan y Peter en 1975 (Tabla 1)1,2,3,5,7, con una sensibilidad y especificidad aproximadamente del 45-90 y del 90%, respectivamente. Con posterioridad, se han desarrollado numerosos criterios de clasificación como los de Tanimoto (1995), que incluyen los síndromes de solapamiento y los anticuerpos anti-Jo1, pero no el resto de anticuerpos ni la miopatía por cuerpo de inclusión8,9.
| Tabla 1. Criterios diagnósticos propuestos por Bohan y Peter en 1975 |
|---|
| 1. Debilidad muscular proximal y simétrica de la cintura escapular y pelviana |
| 2. Elevación de enzimas musculares séricas |
| 3. Cambios electromiográficos |
| 4. Biopsia muscular compatible |
| 5. Lesiones cutáneas características |
|
Diagnóstico definitivo: más de tres criterios con lesiones cutáneas características Diagnóstico probable: dos criterios con lesiones cutáneas características Diagnóstico posible: un criterio con lesiones cutáneas características |

En 2017, se han publicado los nuevos criterios de clasificación por el grupo IMACS (The International Myositis Assessment and Clinical Studies). Incluyen como principal novedad que, en función de si el paciente tiene biopsia compatible o no, se otorga una puntuación diferente y la correspondiente probabilidad para presentar una MIIC concreta. Además, los pacientes con lesiones cutáneas patognomónicas de DM o DMJ no necesitan obligatoriamente biopsia muscular para poder ser clasificados8.
Dentro de las pruebas complementarias, las más usadas son el ENG/EMG, que suele mostrar un patrón miopático con denervación, la RM, y la biopsia muscular2,5,7. La RM muscular es una prueba no invasiva que puede mostrar áreas de inflamación muscular y permite estudiar áreas musculares más amplias2,5. Es útil como control del curso de la enfermedad2,5,8 y puede utilizarse para seleccionar las zonas afectadas donde realizar la biopsia muscular2,5. La biopsia muscular es la prueba definitiva para establecer el diagnóstico, permitiendo valorar la presencia de infiltrado inflamatorio linfohistiocitario, atrofia de predominio perifascicular y necrosis de las fibras musculares1, así como estudiar la sobreexpresión del HLA tipo I en el sarcolema y sarcoplasma de la célula muscular4,5,6. Existen discrepancias sobre la realización de biopsia a todos los pacientes. Algunos expertos recomiendan realizarla a todos los pacientes con DMJ para mejorar la clasificación, el tratamiento y pronóstico a largo plazo4.
Respecto a las pruebas de laboratorio, las enzimas musculares se encuentran elevadas (CK, LDH, aldolasa y AST)1,5, aunque en fases precoces de la enfermedad pueden mantenerse normales. En el 60% de los pacientes pueden detectarse ANA positivos, aunque estos no son específicos de la enfermedad5. En niños también se ha descrito la asociación con anticuerpos anti-Mi-2 (5% de los casos y asociado a buena respuesta al tratamiento), antisintetasa (5-25%, asociado a un curso agudo y con posibles complicaciones como artritis o enfermedad intersticial pulmonar) y anti-p155 o anti-TIF1γ (20-30% de pacientes)1,5,8.
El tratamiento generalmente consiste en la administración de corticoides e inmunosupresores durante un periodo largo aproximado de 1 a 3 años, con el objetivo de alcanzar y mantener la remisión y prevenir las complicaciones5. Inicialmente se pautan corticoides (1-2 mg/kg/día) por vía oral hasta alcanzar una mejoría clínico-analítica y, posteriormente, en dosis descendentes durante 2 años5. No obstante, en la actualidad, muchos pacientes son tratados de forma precoz con inmunosupresores y/o bolos de metilprednisolona (30 mg/kg/día, con un máximo de 1 g/día), lo cual permite una reducción más rápida de los corticoides, con buena respuesta terapéutica1,3,5.
Los inmunosupresores son utilizados como agentes ahorradores de corticoides, siendo el MTX es el más utilizado, administrado en dosis de 10-15 mg/m2 por semana, por vía oral o subcutánea1,5. Para el tratamiento de las manifestaciones cutáneas, se utiliza la hidroxiclorocina1,5 y micofenolato mofetilo3,5.
Las inmunoglobulinas intravenosas (2 g/kg cada 2 semanas, 3 dosis) se emplean en casos de resistencia o dependencia a los corticoides y en enfermedad cutánea resistente y son bien toleradas1,3,5. La ciclofosfamida se plantea en pacientes graves con ulceraciones cutáneas, afectación gastrointestinal y pulmonar3,5.
Existen ensayos con anti-TNFα (infliximab, adalimumab y etanercept) y anti-CD20 (rituximab) con resultados variables3,5.
Como fármacos de segunda línea se ha usado azatioprina y el tacrólimus sistémico y tópico3,5.
La fisioterapia y la terapia ocupacional son de gran importancia para la rehabilitación de los pacientes5.
Un tratamiento insuficiente o el inicio tardío es uno de los predictores más importantes de mal pronóstico, pudiendo aparecer complicaciones como retraso en el crecimiento, calcinosis cutánea o la lipoatrofia2,4,5.
El diagnóstico y tratamiento precoces de la DMJ, por lo tanto, constituyen la clave para mejorar el pronóstico de estos pacientes5.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
ANA: anticuerpos antinucleares · DMJ: dermatosis juvenil · ECG: electrocardiograma · EMG: electromiograma · ENG: electroneurograma · FR: factor reumatoide · MTX: metotrexato · RM: resonancia magnética.
BIBLIOGRAFÍA
- Cabeza Martínez R, Leis Dosil V, Barchino L, Hernanz Hermosa JM. Lesiones cutáneas y debilidad muscular en una niña de 11 años de edad. Acta Pediatr Esp. 2006;64:386-90.
- Corral-Magaña O, Bauzá-Alonso AF, Escudero-Góngora MM, Lacruz L, Martín-Santiago A. La resonancia magnética muscular y el tratamiento agresivo precoz, claves en la dermatomiositis juvenil. Actas Dermosifiliogr. 2018;109:e42-e46.
- Papa V, Romanin B, Bergamaschi R, Cordelli DM, Costa R, De Giorgi LB, et al. Juvenile dermatomyositis: A report of three cases. Ultrastruct Pathol. 2016;40:83-5.
- Iglesias E, Jou C, Bou R, Antón J. Importancia de la biopsia muscular en el diagnóstico de dermatomiositis juvenil. An Pediatr (Barc). 2014;80:e25-e26.
- Bou R, Ricart S. Dermatomiositis juvenil. An Pediatr Contin. 2010;8:183-90.
- Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664-78.
- Phillippi K, Hoeltzel M, Byun Robinson A, Kim S; Childhood Arthritis and Rheumatology Research Alliance (CARRA) Legacy Registry Investigators. Race, Income and Disease Outcomes in Juvenile Dermatomyositis. J Pediatr. 2017;184:38-44.
- Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76:1955-64.
- Tansley SL, Betteridge ZE, Simou S, Jacques TS, Pilkington C, Wood M, et al. Anti-HMGCR Autoantibodies in Juvenile Idiopathic Inflammatory Myopathies Identify a Rare but Clinically Important Subset of Patients. J Rheumatol. 2017;44:488-92.