
Vol. 23 - Num. 91
Notas clínicas
Síndrome de osteoporosis pseudogliomatosa: asociación de ceguera con fracturas múltiples
Juan Ramón y Cajal Calvoa, Ramón Ortiz Giméneza, Marta Zamora Lozanob
aServicio de Radiodiagnóstico. Hospital Clínico Universitario Lozano Blesa. Zaragoza. España.
bSección de Traumatología y Cirugía Ortopédica Infantil. Hospital Clínico Universitario Lozano Blesa. Zaragoza. España.
Correspondencia: J Ramón y Cajal. Correo electrónico: jramonycajalc@gmail.com
Cómo citar este artículo: Ramón y Cajal Calvo J, Ortiz Giménez R, Zamora Lozano M. Síndrome de osteoporosis pseudogliomatosa: asociación de ceguera con fracturas múltiples. Rev Pediatr Aten Primaria. 2021;23:309-12.
Publicado en Internet: 27-09-2021 - Número de visitas: 9302
Resumen
La osteoporosis primaria reúne un compendio de etiologías emergentes y puede ocurrir de forma sindrómica. La asociación de múltiples fracturas óseas con la presencia de pliegues retinianos congénitos debe orientar al diagnóstico del síndrome de osteoporosis-pseudogliomatosa.
Se trata de un trastorno autosómico recesivo poco común, caracterizado por una osteoporosis grave y ceguera de aparición temprana. Las mutaciones de pérdida de función en el gen que codifica la proteína 5 relacionada con el receptor de lipoproteínas de baja densidad (LRP5) parece ser el causante de la aparición de esta enfermedad.
Se presenta un caso de un niño con vítreo hiperplásico bilateral y antecedentes de fracturas cuyo estudio clínico, bioquímico y genético confirman el diagnóstico de esta inusual patología.
Palabras clave
● Ceguera ● Fracturas múltiples ● Genética ● OsteoporosisINTRODUCCIÓN
El síndrome de osteoporosis-pseudogliomatosa es un trastorno autosómico recesivo poco común, caracterizado por una osteoporosis grave y ceguera de aparición temprana. Las mutaciones de pérdida de función en el gen que codifica la proteína 5 relacionada con el receptor de lipoproteínas de baja densidad (LRP5) se han establecido como el defecto genético de la enfermedad1.
CASO CLÍNICO
Se presenta un niño de cuatro años, procedente de Gambia, con antecedentes de múltiples fracturas cuyos padres refieren consanguinidad de segundo grado. El paciente se encuentra en seguimiento en consultas de Endocrinología y Traumatología Pediátrica con el diagnóstico de psteoporosis-pseudogliomatosa (mutación del gen LRP5 confirmada). Refiere una hermana mayor de 15 años con el mismo cuadro clínico e historial médico de numerosas fracturas.
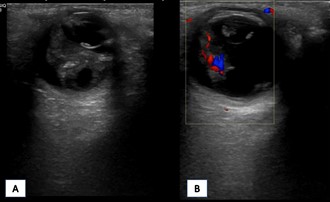
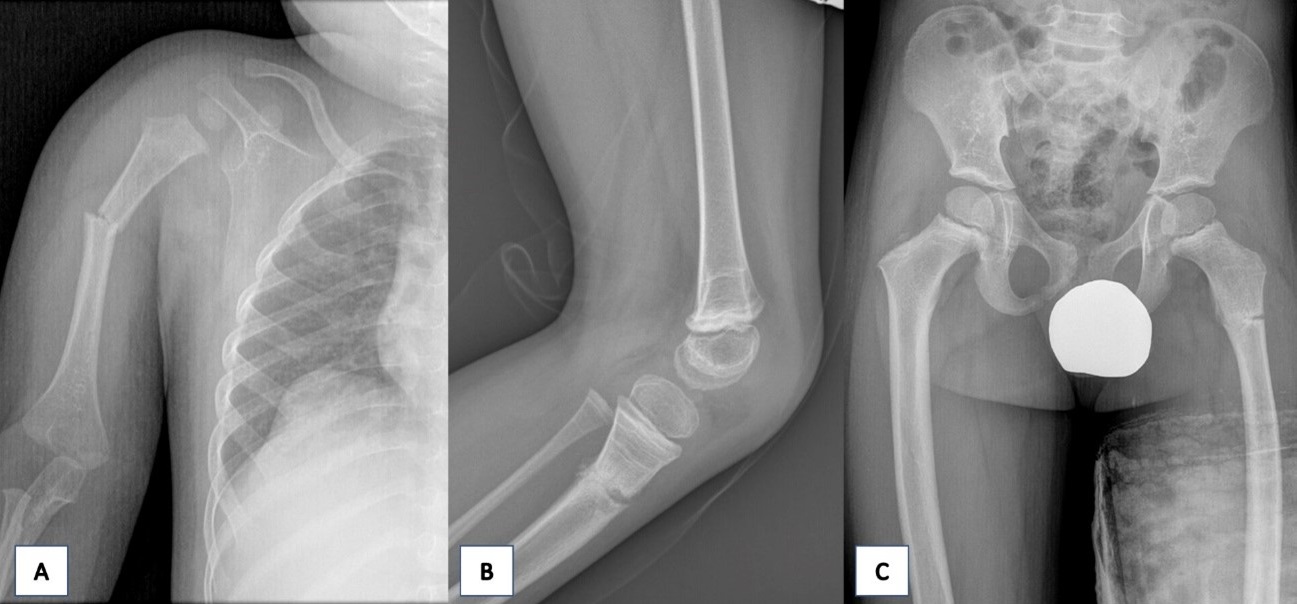
Durante el periodo neonatal, se le diagnóstico de vítreo primario hiperplásico bilateral, que derivaron en un deterioro progresivo e irreversible de la función visual (Fig. 1). A los 2 años de vida, presentó una fractura diafisaria de humero proximal derecho desplazada tras un pequeño golpe mientas jugaba con su hermano mayor, que precisó de reducción ortopédica (Fig. 2A). Al año siguiente, otro traumatismo menor le originó una fractura de la meseta tibial izquierda sin desplazamiento que fue tratado mediante inmovilización y tratamiento conservador (Fig. 2B). A los cuatro años sufrió una caída en bicicleta que le provocó una fractura subtrocantérea del fémur izquierdo (Fig. 2C).
| Figura 1. Ecografía oftálmica. Se visualiza una masa hiperecoica bilateral retrolental que ocupa toda la cámara vítrea desplazando hacia delante el cristalino (A). La masa presenta captación Doppler (B), siendo compatible con la presencia de vítreo primario hiperplásico bilateral. |
|---|
 |
| Figura 2. A. Radiografía anteroposterior del húmero derecho. Se visualiza disrupción y angulación cortical de la diáfisis proximal correspondiente con fractura desplazada. B. Radiografía lateral rodilla izquierda. Se observa una fractura metafisaria de la tibia. C. Radiografía de pelvis anteroposterior: se pone de manifiesto la disrupción y radiolucencia cortical correspondiente con fractura subtrocantérea del fémur izquierdo sin desplazamiento. |
|---|
 |
El paciente recibe en la actualidad tratamiento con calcio más vitamina D e inicio reciente con zolendronato intravenoso de forma programada.
En la exploración física, se constata un peso y talla dentro del percentil 60 con un desarrollo madurativo dentro de los parámetros normales. Los resultados bioquímicos del metabolismo cálcico no evidenciaron resultados significativos. La densitometría ósea de la columna lumbar mostró una DMO L2-L4 de 0,432 g7cm2, puntación Z de -3,6 derivaciones estándar (DE).
Las fracturas múltiples en un niño suelen recordar al diagnóstico de osteogénesis imperfecta, pero también es importante considerar otro posible diagnóstico como es el síndrome de osteoporosis-pseudogliomatosa.
DISCUSIÓN
Este síndrome, descrito por primera vez en el año 1996, es un raro trastorno de herencia autosómica recesiva con una prevalencia de 1/2 000 000, con mayor incidencia en poblaciones que presentan consanguinidad2 y que se caracteriza por tener una variedad de manifestaciones, siendo dos de las más características y constantes la osteopenia generalizada y la regresión vascular ocular fetal.
La característica distintiva de este síndrome es sin duda, la lesión ocular, siendo persistente la afectación grave de las partes coriorretinianas del ojo, microoftalmía, anomalías de la cámara anterior, cataratas, anomalías vitrorretinianas, con discapacidad visual que a menudo conduce a una ceguera completa3.
Las características radiográficas incluyen platispondilia y cifoescoliosis, junto con el arqueamiento de los huesos largos y múltiples fracturas. Otras manifestaciones clínicas descritas son la presencia de microcefalia, hipotonía muscular y retraso mental4.
Se presume que el síndrome de osteoporosis pseudogliomatosa es una enfermedad del tejido conectivo, pero en la actualidad, todavía no se ha encontrado un defecto del metabolismo del colágeno3. Los parámetros bioquímicos del metabolismo fosfocálcico no están alterados en la mayoría de las ocasiones, como en el caso de nuestro paciente.
El tratamiento propuesto en estos momentos reside en una buena alimentación y la promoción de la actividad física con el fin de estimular la estimulación ósea, así como el tratamiento farmacológico basado en la administración de bifosfonatos y vitamina D con una duración aún por determinar5. El proceso osteopénico parece estabilizarse con la edad, por lo tanto, la profilaxis y el cuidado de las fracturas y deformidades son necesarios para prevenir problemas incapacitantes a largo plazo.
En la actualidad, todavía queda mucho por conocer acerca de este trastorno y su fisiopatología, debido a lo cual, resulta imprescindible un abordaje interdisciplinar para realizar un diagnóstico precoz, anticipar posibles comorbilidades y conseguir un tratamiento individualizado a las necesidades del paciente.
CONFLICTO DE INTERESES
Los autores declaran no presentan conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
DE: derivaciones estándar · LRP5: proteína 5 relacionada con el receptor de lipoproteínas de baja densidad.
BIBLIOGRAFÍA
- Papadopoulos I, Bountouvi E, Attilakos A, Gole E, Dinopoulos A, Peppa M, et al. Osteoporosis-pseudoglioma syndrome: clinical, genetic, and treatment-response study of 10 new cases in Greece. Eur J Pediatr. 2019;178:323-9.
- Braslavsky D, Scaglia P, Sanguineti N, Aza-Carmona M. Síndrome de osteoporosis-pseudoglioma: a propósito de un caso pediátrico de osteoporosis primaria. Arch Argent Pediatr. 2020;118:300-4.
- McDowell CL, Moore JD. Multiple fractures in a child: the osteoporosis pseudoglioma syndrome. A case report. J Bone Joint Surg Am. 1992;74:1247-9.
- Balemans W, Van Hul W. The genetics of low-density lipoprotein receptor-related protein 5 in bone: a story of extremes. Endocrinology. 2007;148:2622-9.
- Streeten EA, Ramírez S, Eliades M, Jaumungal S. Fractures on bisphosphonates in osteoporosis pseudoglioma syndrome (OPPG): pQCT shows poor bone density and structure. Bone. 2015;77:17-23.