
Vol. 23 - Num. 90
Casos clínicos en Digestivo
Síndrome de Joubert
Sara Marquina Cintoraa, Ariadna Sánchez Suáreza, Myriam Herrero Álvarezb, Iván Carabaño Aguadoc, Elisa Aguirre Pascuald, Enrique Salcedo Lobatoc, Enrique Medina Benítezc
aServicio de Pediatría. Hospital Universitario de Getafe. Getafe. Madrid. España.
bServicio de Pediatría. Hospital Universitario Rey Juan Carlos. Móstoles. Madrid. España.
cSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
dSección de Radiología Infantil. Servicio de Radiodiagnóstico. Hospital Universitario 12 de Octubre. Madrid. España.
Correspondencia: S Marquina. Correo electrónico: sara.marquina5@gmail.com
Cómo citar este artículo: Marquina Cintora S, Sánchez Suárez A, Herrero Álvarez M, Carabaño Aguado I, Aguirre Pascual E, Salcedo Lobato E, et al. Síndrome de Joubert. Rev Pediatr Aten Primaria. 2021;23:191-4.
Publicado en Internet: 22-06-2021 - Número de visitas: 36392
Resumen
El síndrome de Joubert es una enfermedad multisistémica poco frecuente. Se caracteriza por una malformación congénita del tronco cerebral e hipoplasia del vermis cerebeloso. Estas alteraciones provocan, entre otros, problemas respiratorios, hipotonía y retraso del desarrollo psicomotor. Puede asociar alteraciones a nivel renal, hepático u ocular, entre otros. No existe un tratamiento curativo de la enfermedad; por ello es importante un diagnóstico precoz, para ofrecer un seguimiento multidisciplinar y así poder mejorar el pronóstico y la calidad de vida de los pacientes y sus familias.
Palabras clave
● Cerebelo ● Elastografía ● Fibrosis quística ● JoubertINTRODUCCIÓN
El síndrome de Joubert es una enfermedad neurológica congénita que implica una malformación del tronco del encéfalo y el cerebelo.
Esta enfermedad comienza a expresarse en el periodo neonatal con alteraciones del patrón respiratorio. Posteriormente mejora dicho patrón, y aparecen nuevos síntomas neurológicos, como hipotonía o retraso del desarrollo psicomotor. Se trata de un cuadro clínicamente heterogéneo, que puede asociar alteraciones a nivel renal, hepático u ocular, entre otros. Se deben realizar pruebas complementarias periódicas como parte de la búsqueda activa de estas alteraciones1.
A continuación se describe el caso de un paciente con síndrome de Joubert con afectación hepática en su evolución.
CASO CLÍNICO
Presentamos el caso de un paciente de diez años, con antecedente de síndrome de Joubert, derivado a consultas de Gastroenterología Infantil por afectación hepática.
No presentaba antecedentes neonatales relevantes, salvo una alteración cerebelosa que se visualizó en ecografía en la semana 35 de gestación. Padres consanguíneos (primos).
Fue derivado a Neuropediatría por retraso psicomotor, coloboma retiniano bilateral, apraxia oculomotora e hipotonía severa. Se solicitaron pruebas complementarias, entre ellas una resonancia magnética cerebral, en la que se objetivaron malformaciones en fosa posterior (displasia cerebelosa con alteraciones en vermis y pedúnculos cerebelosos superiores), evocando una imagen del "signo del diente molar".
Ante estos hallazgos, se solicitó estudio genético. Este se informó como cariotipo normal (46XY) y una mutación en homocigosis en el gen TMEM67, descrita previamente asociada al síndrome de Joubert.
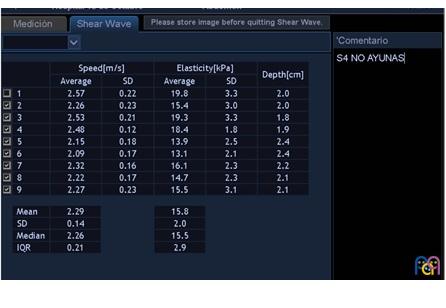
Tras el diagnóstico de síndrome de Joubert, se inició seguimiento multidisciplinar del paciente. Durante el seguimiento posterior, a los 10 años de edad, se detecta en control analítico una hipertransaminasemia creciente con patrón de colestasis sin hiperbilirrubinemia, y con tendencia a la trombopenia. Se deriva a consultas de Gastroenterología del hospital terciario de referencia, para proceder a un despistaje de hipertensión portal. En dicho centro, como primera etapa, se solicitó ecografía abdominal con Doppler y elastografía hepática. Ambas pruebas muestran alteraciones. De especial valor resulta la elastografía, en la que se objetivan unos valores compatibles con aumentado del grado de rigidez (de media, 15,8 kPa) (Figs. 1 y 2). En la ecografía Doppler se constató una hepatomegalia leve, sin alteraciones significativas de los flujos sanguíneos a través de la vena porta, arteria hepática y venas suprahepáticas.
| Figura 1. Ecografía con elastografía. La “rigidez” del hígado evaluado se expone a través de un código de colores |
|---|
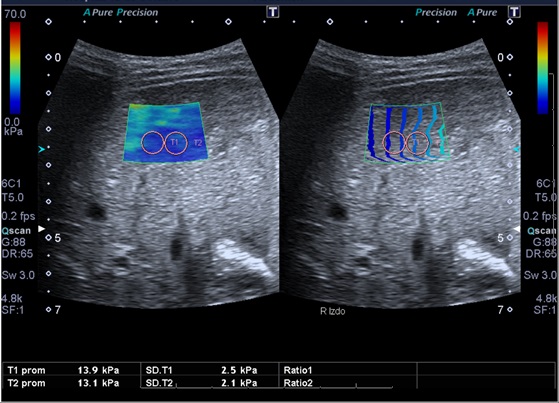 |

| Figura 2. Valores numéricos de elastografía y velocidad de la transmisión ecográfica. A mayor cifra de kPa, mayor será la rigidez del hígado (y, por tanto, mayor será el grado de fibrosis) |
|---|
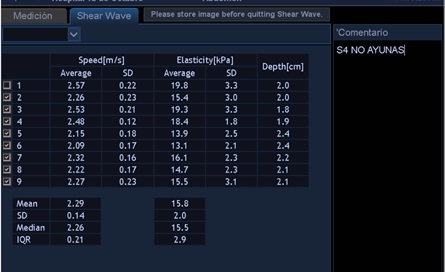 |
DISCUSIÓN
El síndrome de Joubert es una enfermedad genética, neurológica, que fue descrita por primera vez en la literatura, en 1969, por Marie Joubert. Se caracteriza por una malformación del tronco del encéfalo y agenesia o hipoplasia del vermis cerebeloso1. Este síndrome normalmente tiene un patrón de herencia autosómico recesivo, y forma parte de un conjunto de enfermedades catalogadas como ciliopatías congénitas1. No se ha determinado la prevalencia exacta de esta patología, pero muchos autores utilizan un rango entre 1:80 000 y 1:100 000, pudiendo ser una subestimación de las cifras reales1.
El síndrome de Joubert clásico se caracteriza por tres hallazgos principales: malformación del cerebelo y tronco del encéfalo denominada signo del diente molar, hipotonía y retraso del desarrollo. Habitualmente, su manifestación más precoz es la alteración del patrón respiratorio (taquipnea o apnea episódica neonatal), a la que se suelen añadir nistagmo y apraxia oculomotora. Meses después, puede aparecer hipotonía. Más tarde, se hace evidente una ataxia cerebelosa. Los hitos neurológicos se retrasan, y las capacidades cognitivas pueden verse mermadas. Otras anomalías descritas en este contexto: enfermedad renal (nefronoptisis), fibrosis hepática, distrofia retiniana, colobomas oculares, polidactilia, hamartomas orales y alteraciones endocrinas1-6.
Dentro de los subtipos que existen en este síndrome, nos centraremos en el síndrome de Joubert asociado a enfermedad hepática. Existen dos subtipos con estas características: el síndrome Gentile y el síndrome COACH1-6.
La asociación de síndrome de Joubert y fibrosis hepática se denomina síndrome Gentile1. La fibrosis hepática se debe a un trastorno del desarrollo del sistema portobiliar, consistente en una remodelación defectuosa de la placa biliar y una fibrosis progresiva de los tractos portales2. Puede derivar en hepatoesplenomegalia, hipertensión portal, varices esofágicas y cirrosis2,3.
Cuando el síndrome de Joubert se asocia a fibrosis hepática, oligofrenia, ataxia y colobomas, se denomina síndrome hipoplasia del vermis cerebeloso, oligofrenia, ataxia congénita, coloboma y fibrosis hepática (Cerebellar vermis hypo/aplasia, Oligophrenia, Ataxia congenital, Coloboma, and Hepatic fibrosis) (COACH). Se ha detectado la mutación en MKS3 como la principal responsable del síndrome COACH. Además, existen otras mutaciones asociadas, pero mucho menos frecuentes, como la CC2D2A y RPGRIP1L4.
En estos pacientes es importante evaluar, como primer paso, la realización de elastografía hepática, para estimar la rigidez el hígado y, por consiguiente, si puede existir fibrosis o no.
Los grados de fibrosis se dividen en cinco, siendo F0 equivalente a hígado sano y F4, máxima fibrosis o cirrosis. El resultado de la elastografía se expresa en kilopascales (kPa), y se correlaciona con los grados de fibrosis (F0-F4). El rango de equivalencias varía en función del tipo de elastografía que se realice, por lo que estos valores no son comparables entre técnicas elastográficas distintas, pero si el grado de fibrosis en que se clasifiquen1-6. Se trata de una técnica radiológica sencilla y no invasiva, que nos permitirá realizar una monitorización del hígado, permitiendo repetirla periódicamente al tratarse de una exploración segura para el paciente. No obstante, hay que recalcar que el “patrón oro” diagnóstico de la fibrosis hepática es la biopsia.
El diagnóstico del síndrome de Joubert se basa en los datos clínicos (hipotonía, ataxia, apraxia oculomotora y retraso en el desarrollo) y radiológicos. En algunos casos se detecta una mutación genética asociada al síndrome. En la resonancia magnética cerebral se aprecia una imagen muy característica denominada el signo del diente molar. Esta imagen, que para algunos autores es patognomónica5, se forma como consecuencia de la horizontalización de los pedúnculos cerebelosos superiores y la ausencia del vermis cerebeloso.
Respecto a la genética, se han detectado siete genes asociados con esta enfermedad hasta el momento, entre ellos: AHI1 (6q23), NPHP1 (2q13), CEP290 (12q21), TMEM67 (8q22), RPGRIP1L (16q12), ARL13B (3p12.3-q12.3) y CC2D2A (4p15), y dos loci en los cromosomas 9q34 (JBTS1) y 11p12-q13 (CORS2/JBTS2)6.
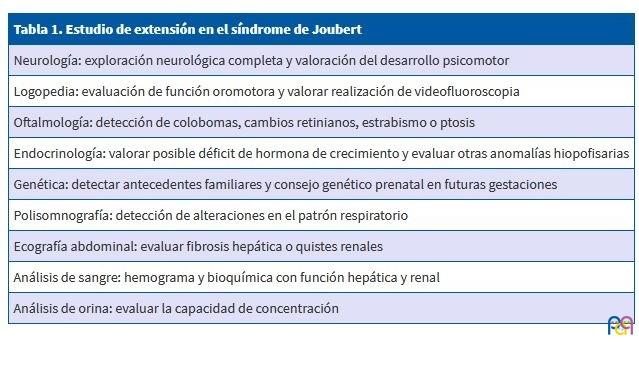
Tras el diagnóstico, para establecer la extensión de la enfermedad, se recomiendan los estudios complementarios que se exponen en la Tabla 1.
| Tabla 1. Estudio de extensión en el síndrome de Joubert |
|---|
| Neurología: exploración neurológica completa y valoración del desarrollo psicomotor |
| Logopedia: evaluación de función oromotora y valorar realización de videofluoroscopia |
| Oftalmología: detección de colobomas, cambios retinianos, estrabismo o ptosis |
| Endocrinología: valorar posible déficit de hormona de crecimiento y evaluar otras anomalías hiopofisarias |
| Genética: detectar antecedentes familiares y consejo genético prenatal en futuras gestaciones |
| Polisomnografía: detección de alteraciones en el patrón respiratorio |
| Ecografía abdominal: evaluar fibrosis hepática o quistes renales |
| Análisis de sangre: hemograma y bioquímica con función hepática y renal |
| Análisis de orina: evaluar la capacidad de concentración |
El tratamiento de la enfermedad es principalmente sintomático y debe ser multidisciplinar. Suele incluir programas de terapia física y adaptación de la educación a los aspectos cognitivos y conductuales del paciente, así como tratamiento de las complicaciones o síntomas asociados que aparezcan. Es importante que el papel del pediatra de Atención Primaria sea integrador y coordine las distintas derivaciones.
El pronóstico es favorable para las formas moderadas de la enfermedad. En las variantes con fibrosis hepática, las complicaciones derivadas de la hipertensión portal marcan el pronóstico.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
Síndrome COACH: hipoplasia del vermis cerebeloso, oligofrenia, ataxia congénita, coloboma y fibrosis hepática (Cerebellar vermis hypo/aplasia, Oligophrenia, Ataxia congenital, Coloboma, and Hepatic fibrosis).
BIBLIOGRAFÍA
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20.
- Angemi JA, Zuccotti JC. Actualizaciones sobre síndrome de Joubert. ALCMEON. 2012;18:25-37.
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat. 2009;30:432-42.
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, et al. Mutations in 3 genes (MKS3, RPGRIP1L, and CC2D2A) cause COACH syndrome/Joubert syndrome with congenital hepatic fibrosis. J Med Genet. 2010;47:8-21.
- Poretti A, Boltshauser E, Valente EM. The molar tooth sign is pathognomonic for Joubert syndrome. Pediatr Neurol. 2014;50:15-6.
- Parisi M, Glass I. Joubert Syndrome. En: Gene Reviews [en línea] [consultado el 17/06/2021]. Disponible en www.ncbi.nlm.nih.gov/books/NBK1325/