
Vol. 23 - Num. 89
Casos clínicos en Digestivo
Hepatomegalia, polifagia e hipertransaminasemia
Clara M.ª Aymerich de Franchescia, Andrea Riego Martínezb, Marta Murillo Hernándezb, Silvia Chumillas Calzadac, David Coca Robinotd, M.ª Isabel Álvarez Morae, Iván Carabaño Aguadof, Enrique Medina Benítezf
aSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Hospital Universitario 12 de Octubre. Madrid. España.
bMIR-Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
cUnidad de Enfermedades Raras. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
dSección de Radiología Infantil. Servicio de Radiodiagnóstico. Hospital Universitario 12 de Octubre. Madrid. España.
eServicio de Genética. Hospital Universitario 12 de Octubre. Madrid. España.
fSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
Correspondencia: CM Aymerich. Correo electrónico: claraymerich@gmail.com
Cómo citar este artículo: Aymerich de Franchesci CM, Riego Martínez A, Murillo Hernández M, Chumillas Calzada S, Coca Robinot D, Álvarez Mora MI, et al. Hepatomegalia, polifagia e hipertransaminasemia. Rev Pediatr Aten Primaria. 2021;23:95-7.
Publicado en Internet: 30-03-2021 - Número de visitas: 15781
Resumen
La glucogenosis es una enfermedad metabólica poco frecuente, producida por un trastorno del metabolismo de los hidratos de carbono. Existen múltiples variantes en función de la enzima implicada, la tipo 9 se produce por una deficiencia de la glucógeno desfoforilasa-cinasa a nivel hepático y se caracteriza por la presencia de hepatomegalia, hipertransaminasemia e hipoglucemia con el ayuno. Se presenta el caso de una lactante de 10 meses, cuyos síntomas guías fueron la distensión abdominal y la hiperfagia. El diagnóstico de glucogenosis se confirmó mediante el estudio genético, objetivándose una mutación en el gen PHKG2, compatible con el diagnóstico de enfermedad de almacenamiento de glucógeno tipo 9C. Se instauró tratamiento sintomático, evitando el ayuno y aumentando la ingesta hidratos de carbono de absorción lenta, con buena evolución clínica.
Palabras clave
● Glucogenosis ● Hepatomegalia ● Hiperfagia ● Hipertransaminasemia ● HipoglucemiaINTRODUCCIÓN
La glucogenosis es una enfermedad producida por un error innato del metabolismo de los hidratos de carbono. Hay distintos tipos, cada uno de los cuales presenta rasgos comunes, si bien existen características divergentes. La tipo 9 se produce por una deficiencia de la glucógeno desfoforilasa-kinasa a nivel hepático. Se caracteriza por hepatomegalia, elevación de transaminasas e hipoglucemia con el ayuno. El gold standard para el diagnóstico es la biopsia hepática; cobrando cada vez más importancia el estudio genético.
A continuación, se describe un caso con una forma de presentación característica.
CASO CLÍNICO
Niña de diez meses derivada a consultas de Digestivo por distensión abdominal. Primera hija de padres consanguíneos (primos hermanos), sin antecedentes familiares ni personales de interés. Desde los seis meses de vida, comienza con distensión abdominal progresiva sin otros síntomas. Alimentada con lactancia materna hasta los nueve meses, introducción de alimentación complementaria a los seis meses y fórmula de inicio a partir de los ocho meses, bien tolerada. Desde el punto de vista neurológico, destaca la adquisición ligeramente tardía de la sedestación autónoma, a los diez meses.
En la anamnesis, llama la atención la hiperfagia. Los padres refieren que come con mucho apetito y tiene hambre casi constantemente. También, la irritabilidad nocturna y la tendencia diaforética. En la exploración física, presenta aspecto macrosómico, con prominente panículo adiposo. Buena coloración de piel y mucosas, no ictericia. Abdomen distendido con hepatomegalia masiva de consistencia firme, hasta 12 cm bajo el reborde costal. No hay esplenomegalia. Exploración neurológica normal. En los análisis de sangre, presenta hipoglucemia (20 mg/dl) y elevación de transaminasas (GPT 289 U/l, GOT 550 U/l, GGT 125 U/l), hipertrigliceridemia (240 mg/dl) e hipercolesterolemia (280 mg/dl), creatin cinasa (CK) 89 U/l, lactato 1,3 mmol/l; con estudio de virus hepatotropos y autoinmunidad negativos. Estudio de coagulación normal. En la ecografía abdominal se visualiza una hepatomegalia masiva, con hiperecogenicidad difusa (Figs. 1 y 2). Ecocardiografía y exploración oftalmológica normales. Durante el ingreso, se llevan a cabo controles glucémicos, constatándose numerosas hipoglucemias de ayuno, todas ellas asintomáticas. Ante la sospecha de glucogenosis, se instaura tratamiento dietético con optimización de la ingesta de hidratos de carbono de absorción lenta, con lo que se consigue controles de glucemia normales previos al alta. Se realiza estudio metabólico sugerente de glucogenosis (oligosacáridos en orina: excreción elevada de hex4). En el estudio genético se objetiva variante patogénica c.556G>T(p.E186*) en el gen PHKG2 en aparente homocigosis, compatible con el diagnóstico de enfermedad de almacenamiento de glucógeno tipo 9C.
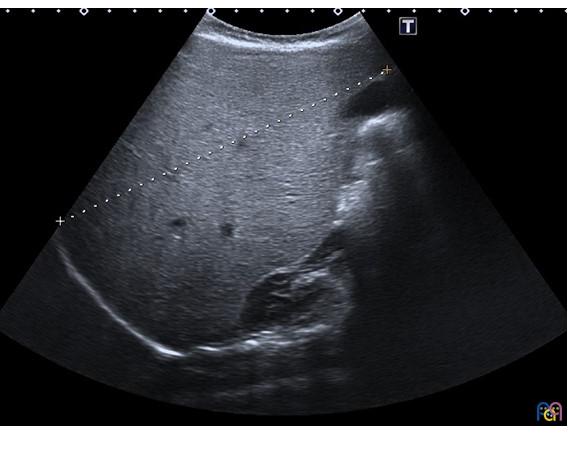
| Figura 1. Gran hepatomegalia con hiperecogenicidad difusa. Se muestra el eje longitudinal del hígado (14,3 cm), muy aumentado de tamaño para la edad de la menor |
|---|
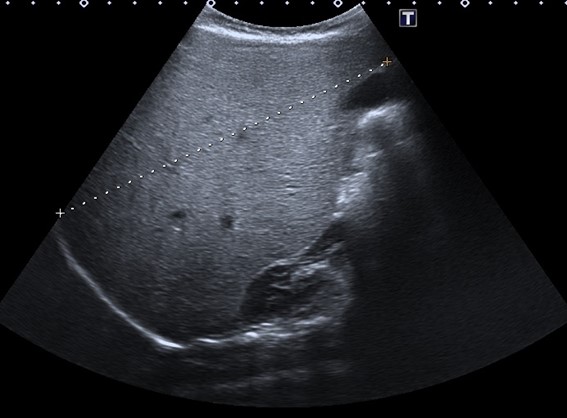 |
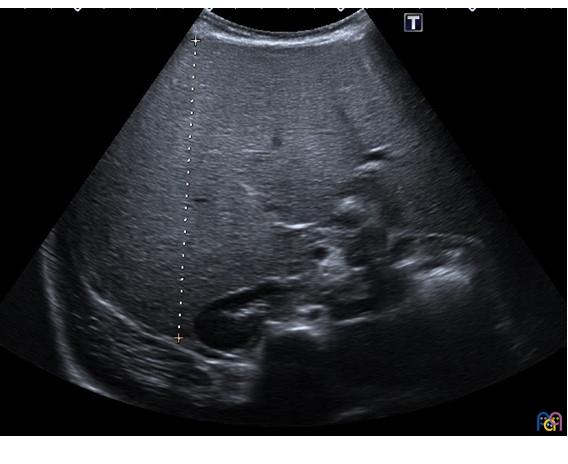
| Figura 2. Eje anteroposterior del hígado (11,2 cm), también muy aumentado de tamaño para la edad de la menor, en un contexto de hiperecogenicidad difusa |
|---|
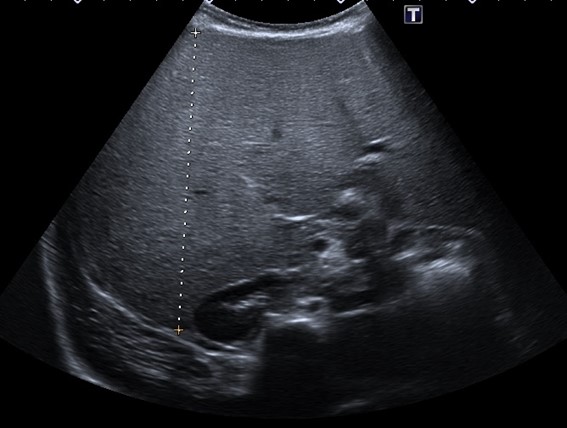 |
DISCUSIÓN
La glucogenosis es una enfermedad poco frecuente, con una incidencia variable según los estudios (1/20 000-60 000 de recién nacidos vivos), por ello es fundamental tener una alta sospecha diagnóstica. Existen varios subtipos de glucogenosis en función de la enzima afectada. En concreto, la tipo 9 se produce por el déficit de la glucógeno fosforilasa-cinasa hepática. El déficit de esta enzima impide la degradación de glucógeno, lo que se traduce en un acúmulo de glucógeno en el hígado y una disminución de glucosa en sangre. Se han descrito varias mutaciones ligadas al cromosoma X y otras de tipo autosómica recesivas. Clínicamente, se caracteriza por hepatomegalia masiva sin colestasis ni esplenomegalia. En nuestro caso, el síntoma guía fue la hiperfagia como reflejo de la hipoglucemia que presentaba en ayunas. Este hecho ha de ser tenido en consideración ya desde las consultas de Atención Primaria, pues fue un hecho muy llamativo. En los análisis, se objetiva hipertransaminasemia, hipoglucemia y cetosis con el ayuno; también, hipercolesterolemia, con lactato y CK en rangos de normalidad.
Existen múltiples herramientas diagnósticas: determinación de actividad enzimática y clásicamente la biopsia hepática. Cada vez cobra mayor importancia la genética que nos ofrece el diagnóstico definitivo de la enfermedad.
El tratamiento es sintomático, la dieta es el pilar fundamental. Deben evitar el ayuno e ingerir hidratos de carbono de absorción lenta de forma frecuente para evitar hipoglucemias. Los aportes de glucosa necesarios son variables en función de la edad.
En cuanto al pronóstico, suele mejorar con la edad, siendo la mayoría asintomáticos en la edad adulta, aunque un pequeño porcentaje desarrollan cirrosis hepática durante la adolescencia.
En conclusión, la glucogenosis es una enfermedad poco frecuente que suele presentarse en la infancia en forma de hepatomegalia masiva e hipoglucemias con el ayuno. No debemos olvidar este diagnóstico ante un niño con hambre voraz y hepatomegalia. Es importante diagnosticarlo precozmente ya que se ha objetivado que una intervención precoz mejora la curva ponderal y hepatomegalia.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflicto de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
CK: creatin cinasa.
BIBLIOGRAFÍA
- Soler P, Tomasa N, Sánchez J, Yeste D, Gussinyé M, Carrascosa A, et al. Hepatomegalia, distensión abdominal e hipoglucemia en un lactante: expresión clínica de la glucogenosis tipo IX. An Pediatr (Barc). 2004;61:438-41.
- Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B, Bali DS, et al. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:772-89.
- Bali DS, Goldstein JL, Fredrickson K, Austin S, Pendyal S, Rehder C, et al. Clinical and Molecular Variability in Patients with PHKA2 Variants and Liver Phosphorylase b Kinase Deficiency. JIMD Rep. 2017;37:63-72.
- Bali DS, Goldstein JL, Fredrickson K, Rehder C, Boney A, Austin S, et al. Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene. Mol Genet Metab. 2014;111:309-13.