
Vol. 23 - Num. 89
Notas clínicas
Displasia torácica asfixiante
Claudia García Barrionuevoa, Pedro Luis Márquez Miraa, Borja Croche Santanderb, Román Jesús López Díazc
aMIR-Pediatría. Hospital Juan Ramón Jiménez. Huelva. España.
bServicio de Pediatría. Hospital Juan Ramón Jiménez. Huelva. España.
cPediatra. CS San Juan del Puerto. Huelva. España.
Correspondencia: C García. Correo electrónico: claudiagarciabarrionuevo@gmail.com
Cómo citar este artículo: García Barrionuevo C, Márquez Mira PL, Croche Santander B, López Díaz RJ. Displasia torácica asfixiante. Rev Pediatr Aten Primaria. 2021;23:91-4.
Publicado en Internet: 29-03-2021 - Número de visitas: 14874
Resumen
El síndrome de Jeune o displasia torácica asfixiante es una enfermedad genética de herencia autosómica recesiva, que se caracteriza por presentar un fenotipo característico. El diagnóstico es clínico y radiológico. Se han descrito mutaciones en genes (IFT80, DYNC2H1, WDR19, IFT140 y TTC21B) que codifican proteínas de transporte intraflagelares responsables de la enfermedad.
Palabras clave
● Malformación congénita ● Síndrome de JeuneINTRODUCCIÓN
El síndrome de Jeune es una enfermedad caracterizada por un tórax estrecho campaniforme, costillas horizontalizadas, clavículas elevadas, huesos largos e iliacos cortos, techo acetabular en forma de tridente, falanges de manos y pies hipoplásicas con epífisis en forma de cono y ocasionalmente polidactilia. Aunque el diagnóstico de sospecha se establece en base a criterios clínicos y radiológicos, la certeza diagnóstica se obtiene cuando se hallan mutaciones en diferentes genes que codifican proteínas de transporte intraflagelares (IFT80, DYNC2H1, WDR19, IFT140 y TTC21B) responsables del desarrollo de la enfermedad1.
CASO CLÍNICO
Presentamos el caso de un niño de cinco años de origen magrebí, hijo de padres consanguíneos, afecto de síndrome de Jeune o displasia torácica asfixiante.
Entre sus antecedentes destaca: embarazo controlado, con diabetes gestacional, polihidramnios y diagnóstico mediante ecografía de huesos largos cortos. Nace a las 37 + 4 semanas de edad gestacional mediante parto eutócico, con peso adecuado para la edad gestacional. Apgar 2/7, precisando reanimación tipo II. Ingresa en cuidados intensivos neonatales por dificultad respiratoria moderada que precisa de soporte con ventilación mecánica no invasiva (CPAP) durante 18 días. Tras retirada de la CPAP, sigue precisando oxigenoterapia suplementaria para mantener adecuadas saturaciones con oxígeno en gafas nasales a 0,2-0,5 lpm, permaneciendo incluso con estas necesidades una vez dado de alta a domicilio. Durante su ingreso se observa fenotipo peculiar en el que destaca tórax estrecho, costillas y fémur corto, y frente abombada. Ante estos hallazgos se realiza mapa óseo que pone de manifiesto tórax estrecho con acortamiento de costillas, acortamiento e incurvación de ambos fémures y alteración de la morfología de palas iliacas. Se establece diagnóstico de sospecha de displasia torácica asfixiante y se realiza estudio de extensión con ecocardiografía, ecografía abdominal y fondo de ojo que son normales.
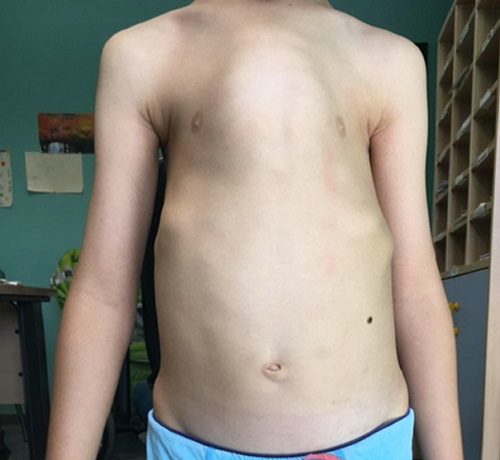
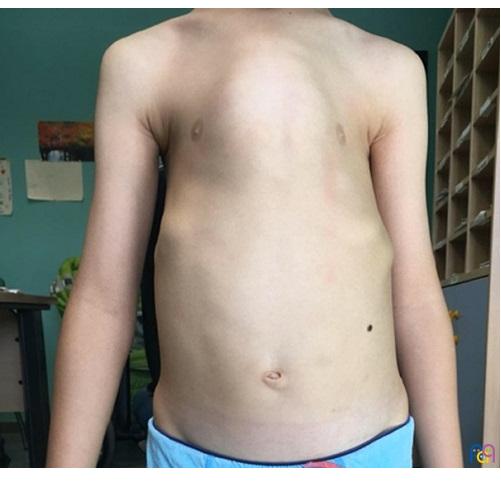
A los cinco años es derivado desde Atención Primaria para valoración de función cardiaca y pulmonar. El paciente presenta un cuadro sindrómico caracterizado por retraso generalizado del desarrollo, estancamiento ponderal y fenotipo peculiar con aspecto macrocéfalo, tórax estrecho con prominencia del esternón (Fig. 1) y de las uniones condrocostales, además de costillas cortas horizontalizadas. El paciente está eupneico, normocoloreado, sin oxigenoterapia para mantener adecuadas saturaciones y realiza una vida normal. En la auscultación se evidencia un soplo sistólico II/VI en borde paraesternal izquierdo sin aparente repercusión hemodinámica. Se solicita tomografía computarizada (TC) de tórax, donde se constata un estrechamiento de la caja torácica en su mitad anterosuperior sin alteraciones en el parénquima pulmonar y cierto acortamiento de costillas craneales, datos compatibles con una displasia esquelética. Ante estos hallazgos y teniendo en cuenta sus antecedentes se solicita estudio genético donde se detecta variante en homocigosis del gen DYNC2H1, que confirma el diagnóstico de síndrome de Jeune.
| Figura 1. Tórax campaniforme |
|---|
 |
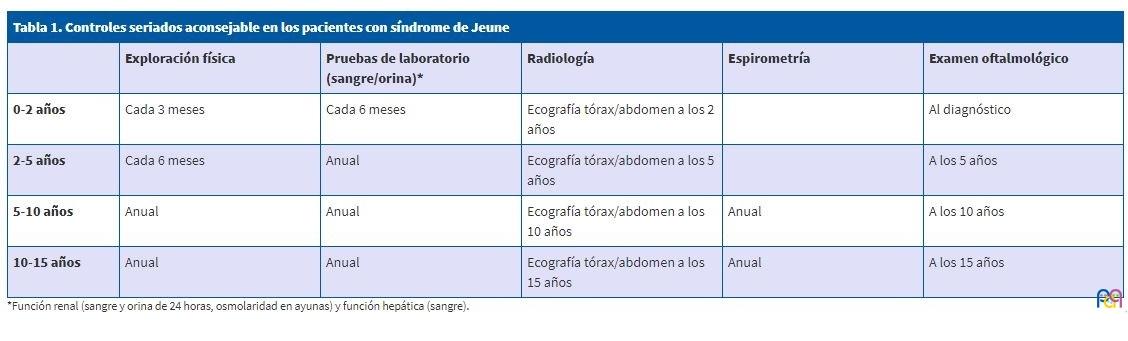
En la actualidad mantiene seguimiento ambulatorio en consultas por las siguientes especialidades (Tabla 1): Neurología, por retraso global del desarrollo; recibe terapia en atención temprana con evolución favorable; Neumología, no realiza tratamiento ni precisa de oxígeno suplementario; Cardiología: corazón estructural y funcionalmente normal; Urología: intervenido de orquidopexia izquierda.
| Tabla 1. Controles seriados aconsejable en los pacientes con síndrome de Jeune | |||||
|---|---|---|---|---|---|
| Exploración física | Pruebas de laboratorio (sangre/orina)* | Radiología | Espirometría | Examen oftalmológico | |
| 0-2 años | Cada 3 meses | Cada 6 meses | Ecografía tórax/abdomen a los 2 años | Al diagnóstico | |
| 2-5 años | Cada 6 meses | Anual | Ecografía tórax/abdomen a los 5 años | A los 5 años | |
| 5-10 años | Anual | Anual | Ecografía tórax/abdomen a los 10 años | Anual | A los 10 años |
| 10-15 años | Anual | Anual | Ecografía tórax/abdomen a los 15 años | Anual | A los 15 años |
DISCUSIÓN
La distrofia torácica asfixiante o síndrome de Jeune es un trastorno genético potencialmente letal. Se hereda de forma autosómica recesiva2. Se ha asociado a diferentes alteraciones genéticas siendo una de ellas la mutación en el gen DYNC2H3. El diagnóstico se basa en hallazgos clínicos y radiológicos4.
La distrofia torácica asfixiante muestra una amplia variabilidad fenotípica y los casos se han clasificado en formas letales, graves, leves y latentes5. La forma clásica de manifestación es tórax estrecho (Fig. 1), palas iliacas cortas y cuadradas, y acortamiento de los miembros, predominantemente el segmento proximal de la extremidad2,6. Los huesos de las extremidades más frecuentemente afectados son cúbito, radio, fémur y tibia5. Pueden presentar polidactilia de manos y pies4,5. La columna vertebral y el cráneo no suelen verse alterados en este síndrome2.
Aunque la mayoría de los casos se determinan por las características esqueléticas, hallándose en más del 70% de los pacientes, existe un amplio espectro de gravedad y variedad en la presentación de los signos2,6.
El compromiso respiratorio se ve afectado en función de la hipoplasia pulmonar. Numerosos recién nacidos con tórax estrecho presentan un grado variable de insuficiencia respiratoria secundaria a la hipoplasia pulmonar, que suele ser grave y letal2,5. Por el contrario, podemos encontrar pacientes que solo manifiesten hipoventilación o infecciones respiratorias recurrentes6. Nuestro paciente presentó al nacimiento dificultad respiratoria sin compromiso vital.
Se han descrito trastornos en diferentes órganos asociados a este síndrome.
- A nivel cardiaco pueden desarrollar insuficiencia cardiaca secundaria al aumento de las resistencias vasculares pulmonares como consecuencia de la constricción torácica e hipoplasia alveolar2.
- A nivel renal se han descrito los siguientes hallazgos: quistes renales, atrofia tubular e insuficiencia renal que pueden aparecer durante la infancia, adolescencia o segunda década de la vida2,6. La afectación renal es la causa predominante de muerte en pacientes desde los tres a los diez años7.
- Las alteraciones hepáticas acontecen en menos del 30% de los casos reportados, resulta difícil predecir el riesgo de desarrollar afectación hepática en función de los hallazgos esqueléticos6.
- A nivel retiniano se han descrito pacientes con distrofia retiniana o anormalidades en la pigmentación de la retina6,8.
- Desde el punto de vista neurológico, cabe la posibilidad de manifestarse como agenesia de cuerpo calloso y síndrome de Dandy-Walker2,6.
- Además, existen casos de malabsorción intestinal, el páncreas puede mostrar cambios quísticos e insuficiencia exocrina6.
- Resulta de gran importancia realizar un diagnóstico diferencial con todas aquellas displasias esqueléticas que cursan con tórax pequeño y costillas cortas.
El pronóstico de este síndrome es difícil de establecer, ya que las complicaciones respiratorias y renales, determinantes en la evolución de los pacientes, no guardan una correlación directa con la severidad de los cambios esqueléticos2,6.
El tratamiento del síndrome de Jeune va dirigido hacia el control de los síntomas y prevención de las complicaciones en la medida de lo posible. La malformación torácica tiende a ser menos severa con la edad6.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
CPAP: ventilación mecánica no invasiva · TC: tomografía computarizada.
BIBLIOGRAFÍA
- Sindrome de Jeune. En: Orphanet [en línea] [consultado el 25/03/2021]. Disponible en www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=474
- Díaz S, Madrid F, León J. PDisplasia torácica asfixiante o síndrome de Jeune. Rev Chile Obst Ginecol. 2003;68:322-6.
- Dagoneau N. DYNC2H1 gene mutations cause asphyxiating thoracic dystrophy and short ribs and polydactyly (SRPS acronym in English) type III syndrome. Am J Hum Genet. 2009;84:706-11.
- De Vries J. Jeune syndrome: description of 13 cases. Monitoring Protocol. Eur J Pediatr- 2010;169:77-88.
- Morgan NV, Bacchelli C, Gissen P. A locus for asphyxiating thoracic dystrophy, ATD, maps to chromosome 15q13. J Med Genet. 2003;40:431-5.
- Keppler-Noreuil KM, Adam MP, Welch J, Muilenburg A, Willing MC. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy). Am J Med Genet A. 2011;155:1021-32.
- Poyner SE, Bradshaw WT. Jeune syndrome: considerations for management of asphyxiating thoracic dystrophy. Neonatal Netw. 2013;32:342-52.
- Casteels I, Demandt E, Legius E. Visual loss as a presenting sign of Jeune syndrome. Eur J Paediatr Neurol. 2000;4:243-7.