
Vol. 22 - Num. 88
Notas clínicas
No es psoriasis todo lo que parece
Nerea Barrado Solísa, Nora Sivó Díazb, Juan Ignacio Marí Ruiza, Carmen Ortega Monzóa
aServicio de Dermatología. Hospital de la Ribera. Alzira. Valencia. España.
bServicio de Pediatría. Hospital de la Ribera. Alzira. Valencia. España.
Correspondencia: N Barrado. Correo electrónico: nerba_87@hotmail.com
Cómo citar este artículo: Barrado Solís N, Sivó Díaz N, Marí Ruiz JI, Ortega Monzó C. No es psoriasis todo lo que parece. Rev Pediatr Aten Primaria. 2020;22:403-6.
Publicado en Internet: 09-12-2020 - Número de visitas: 11700
Resumen
La pitiriasis rubra pilaris (PRP) es una enfermedad cutánea crónica caracterizada por pápulas foliculares, placas anaranjadas que dejan zonas de piel respetada e hiperqueratosis palmoplantar; su edad de presentación y su clínica son muy variables. Se trata de una enfermedad poco frecuente que puede ser mal diagnosticada de otras dermatosis con apariencia similar. Su tratamiento no está bien establecido y a veces puede resultar poco exitoso. Presentamos el caso de una niña de siete años con una clínica típica de PRP y realizamos una breve revisión de esta patología.
Palabras clave
● Pitiriasis rubra pilaris ● PsoriasisINTRODUCCIÓN
La pitiriasis rubra pilaris (PRP) es una dermatosis infrecuente en los niños, que se caracteriza por la presencia de pápulas foliculares, placas de color salmón que dejan islotes de piel respetada y queratodermia palmoplantar. Su diagnóstico en muchas ocasiones supone un reto para el facultativo por su gran parecido con la psoriasis. No obstante, existen ciertas claves en el examen clínico que nos ayudan a sospechar esta enfermedad.
CASO CLÍNICO
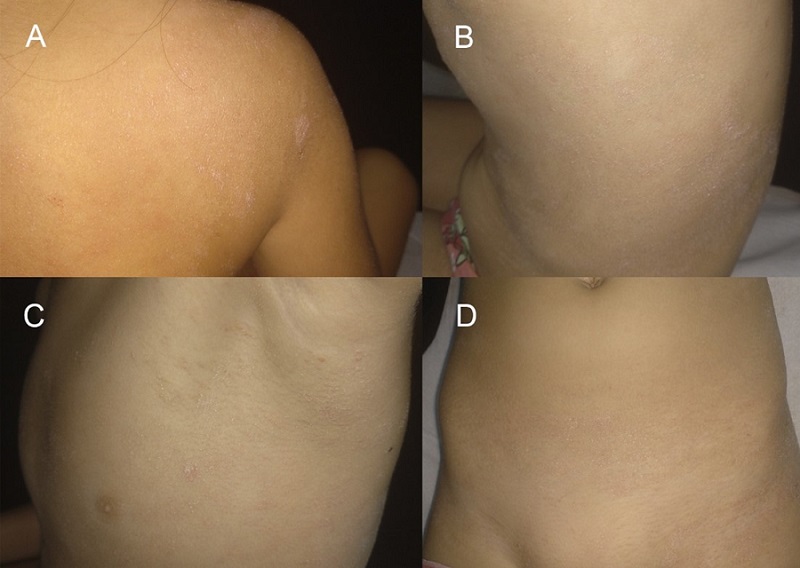
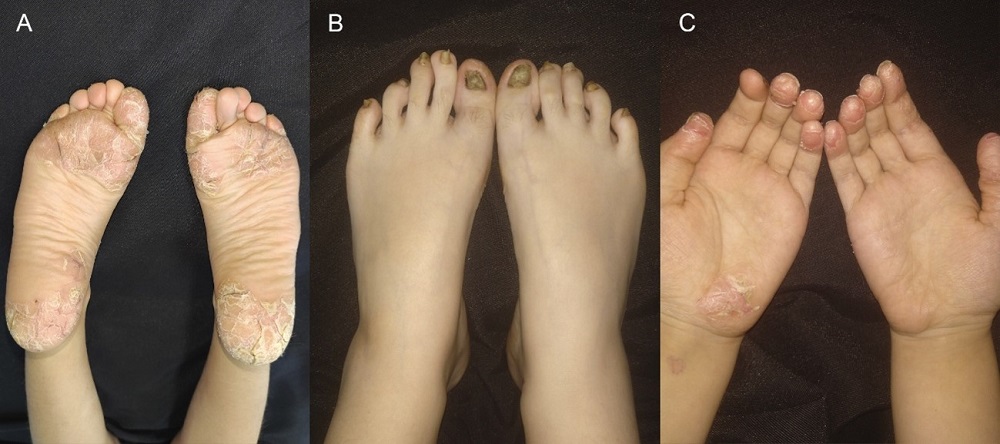
Niña de siete años, sin antecedentes personales ni familiares de interés. Consultó por lesiones eritemato-descamativas pruriginosas generalizadas de dos años de evolución, con mala respuesta a múltiples corticoides tópicos. En la exploración, observamos placas eritematosas y anaranjadas con descamación fina y pápulas foliculares en su interior, que afectaban extensamente al tronco, las extremidades, la zona genital y el cuero cabelludo. Llamaba la atención su distribución en parches, que dejaban áreas de piel respetadas (Fig. 1). Presentaba además placas hiperqueratósicas bien delimitadas, con grietas dolorosas en palmas y plantas, descamación en los pulpejos de los dedos y una intensa afectación ungueal con hiperqueratosis e importante distrofia (Fig. 2).
| Figura 1. Placas extensas eritematoanaranjadas con descamación fina que dejan áreas de piel respetadas y con pápulas foliculares en su interior |
|---|
 |
| Figura 2. Placas hiperqueratósicas palmoplantares con grietas e hiperqueratosis en las uñas de los pies |
|---|
 |

Se realizó una biopsia cutánea de una lesión del tronco que mostró hallazgos compatibles con una PRP. Se inició tratamiento con acitretino a dosis de 0,5 mg/kg tras realizarse un estudio óseo basal, con una espectacular mejoría clínica inicial. A los meses, la paciente volvió a presentar lesiones extensas, generalizadas, así como grietas incapacitantes en las plantas de los pies. Se suspendió acitretino y se introdujo metotrexato, a dosis de 0,3 mg/kg, con una respuesta parcial en la actualidad.
DISCUSIÓN
La PRP es una enfermedad pápulo-escamosa poco frecuente y de etiología desconocida. Es más frecuente en la primera y en la quinta décadas de la vida, no existiendo diferencias según el sexo. Se han propuesto 6 tipos de PRP, según la edad de inicio, la distribución y la apariencia clínica1. Las variantes III, IV y V corresponden a las de presentación pediátrica. Nuestra paciente presenta la tipo III, también llamada PRP clásica juvenil, que se inicia durante la infancia, tiene afectación cutánea generalizada y una tendencia a la resolución espontánea en un periodo de 1-2 años. Clínicamente cursa con placas de color asalmonado que contienen pápulas foliculares y dejan islotes de piel respetados, y se acompaña de queratodermia palmoplantar. La tipo IV suele aparecer en adolescentes y muestra lesiones similares pero circunscritas en codos y rodillas; y la tipo V, o PRP atípica juvenil, también se inicia en la adolescencia y se caracteriza por lesiones generalizadas de ictiosis junto a queratodermia palmoplantar.
Se debe realizar un diagnóstico diferencial con dermatitis atópica, ictiosis, queratosis folicular, liquen espinuloso y especialmente con la psoriasis. Los islotes de piel respetada, la queratodermia palmoplantar y la queratosis folicular en el interior de las placas son claves que orientan a PRP2, aunque normalmente es necesario realizar una biopsia para confirmar el diagnóstico. En cuanto al tratamiento, no existen pautas establecidas y la evidencia existente se basa en series de casos publicados3. La terapia de primera línea son los retinoides orales, acitretino e isotretinoína, cuya respuesta suele ser evidente tras 3-6 meses de tratamiento. En la población infantil los retinoides a altas dosis pueden producir hiperostosis y cierre prematuro de la epífisis, especialmente en pacientes prepuberales4. Por ello se recomienda realizar un estudio de serie ósea previo al tratamiento y repetir cada 12-18 meses, así como buscar la menor dosis y el menor tiempo de tratamiento posible. El metotrexato es considerado un fármaco de segunda línea5. Se utiliza en pacientes que no responden a los retinoides o si estos están contraindicados, siendo su tasa de respuesta inferior. En los últimos años además se han utilizado fármacos biológicos (anti-TNF, anti-IL12-23 y anti-IL17) para aquellos casos más graves y resistentes. Por último, es importante acompañar la terapia sistémica con el uso abundante de emolientes, especialmente aquellos que contienen urea, y se utilizan antihistamínicos y corticoides tópicos de mediana y alta potencia para el alivio sintomático.
CONCLUSIÓN
La PRP es una enfermedad cutánea poco frecuente, pero con una clínica peculiar, que es importante conocer para no confundir con otras patologías papuloescamosas similares como la psoriasis. Presentamos el caso de una niña con una clínica típica de PRP, con lesiones altamente incapacitantes y con pobre respuesta terapéutica.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflicto de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
PRP: pitiriasis rubra pilaris.
BIBLIOGRAFÍA
- Stacey SK, Novek SJ, Maddox CL. Pityriasis Rubra Pilaris in a 3-Year-Old Male. Mil Med. 2016;181:e298-301.
- Wang D, Chong VC, Chong WS, Oon HH. A Review on Pityriasis Rubra Pilaris. Am J Clin Dermatol. 2018;19:377-90.
- Roenneberg S, Biedermann T. Pityriasis rubra pilaris: algorithms for diagnosis and treatment. J Eur Acad Dermatol Venereol. 2018;32:889-98.
- Azaña Defez JM, Martínez Martínez ML. Psoriasis en la infancia y adolescencia. Pediatr Integral. 2016;XX:234-43.
- Alazemi A, Balakirski G, AlShehhi F, Lehmann S, Tenbrock K, Megahed M. Juvenile pityriasis rubra pilaris: successful treatment with methotrexate. Clin Exp Dermatol. 2018;43:110-2.