
Vol. 20 - Num. 79
Notas clínicas
Crisis convulsivas y alteraciones cutáneas: presentación de un caso de esclerosis tuberosa
Paula García Sáncheza, María El Kadaoui Calvob, Blanca Sáez Gallegoc
aServicio de Urgencias. Hospital Universitario Infantil La Paz. Madrid. España.
bPediatra. CS Arroyo de la Media Legua. Madrid. España.
cMIR-Pediatría. Servicio de Pediatría. Hospital Universitario La Paz. Madrid. España.
Correspondencia: P García. Correo electrónico: paula.garsa@gmail.com
Cómo citar este artículo: García Sánchez P, El Kadaoui Calvo M, Sáez Gallego B. Crisis convulsivas y alteraciones cutáneas: presentación de un caso de esclerosis tuberosa. Rev Pediatr Aten Primaria. 2018;20:253-6.
Publicado en Internet: 05-09-2018 - Número de visitas: 15313
Resumen
La esclerosis tuberosa es un síndrome neurocutáneo caracterizado por el desarrollo de hamartomas en distintos órganos. Presenta una herencia autosómica dominante, aunque más del 60% de los casos se deben a mutaciones de novo. La presentación clínica es muy variable, incluyendo principalmente manifestaciones cutáneas, neurológicas, renales, cardiacas y oculares. El diagnóstico se establece por criterios clínicos y estudio genético. El tratamiento consiste en control de la sintomatología neurológica y de otras manifestaciones sistémicas. Presentamos el caso de una niña de cuatro años remitida a Urgencias por crisis convulsivas de corta duración durante una semana, con hallazgo en el examen físico de lesiones cutáneas típicas de esclerosis tuberosa. Las pruebas de neuroimagen revelaron los característicos tuber cerebrales. Se realiza una breve revisión de esta enfermedad.
Palabras clave
● Convulsiones ● Esclerosis tuberosa ● HamartomaCASO CLÍNICO
Niña de cuatro años y siete meses originaria de China remitida a Urgencias por su pediatra por presentar desde hace una semana episodios de desconexión de unos diez segundos de duración, con mirada fija y movimiento de cabeceo. En una ocasión presentó movimientos tónico-clónicos de miembro superior izquierdo durante 15 segundos. Recuperación espontánea sin periodo poscrítico. Afebril, sin otra sintomatología. Antecedentes personales y familiares desconocidos (adopción hace dos meses).
En el examen físico presenta máculas acrómicas en el tronco y las extremidades, angiofibromas y colagenomas faciales, además de una característica placa de Chagrin en espalda. La exploración neurológica es normal, sin datos de focalidad. Crecimiento y desarrollo adecuados para su edad (peso en p45 y talla en p30).
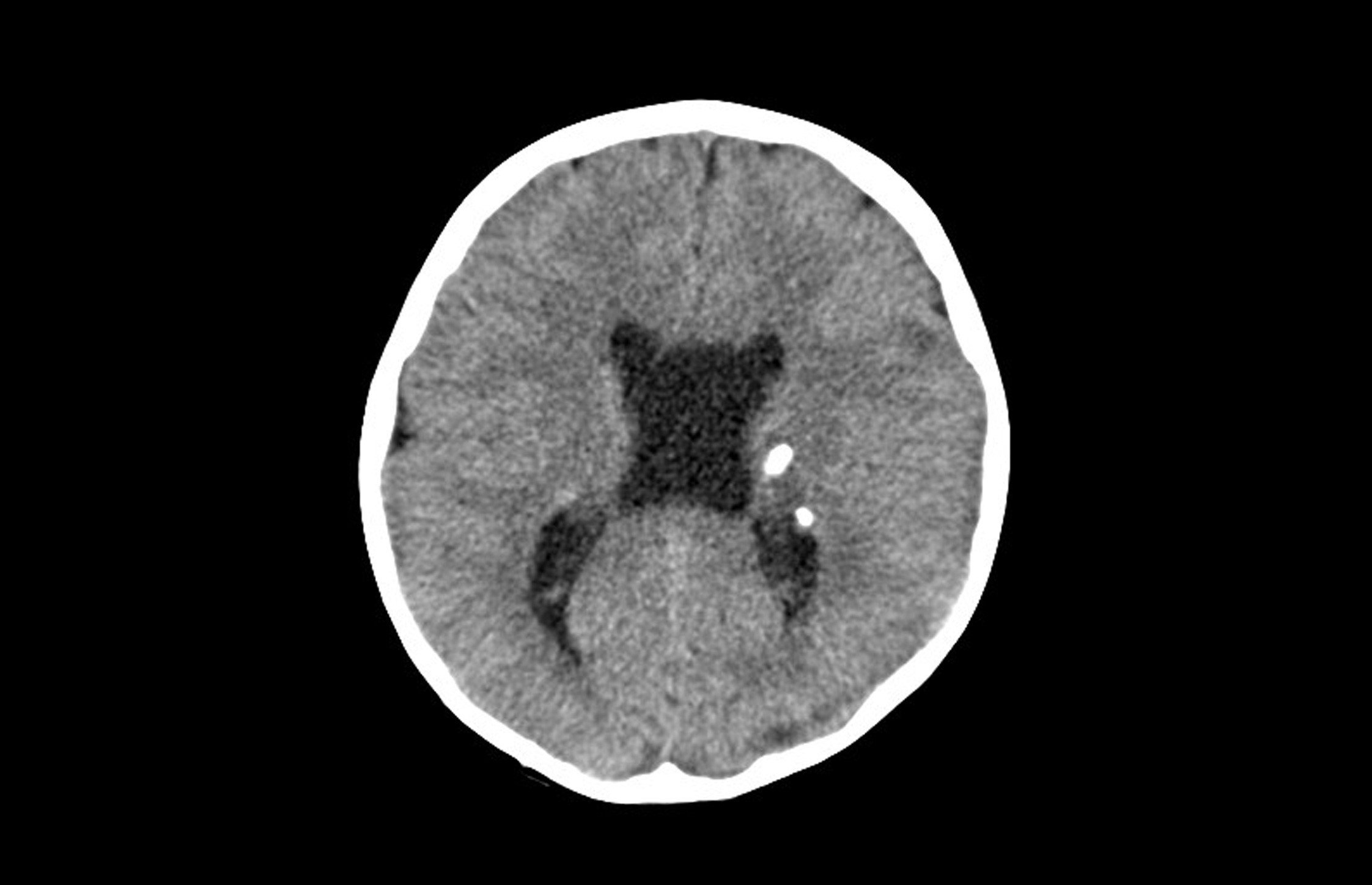
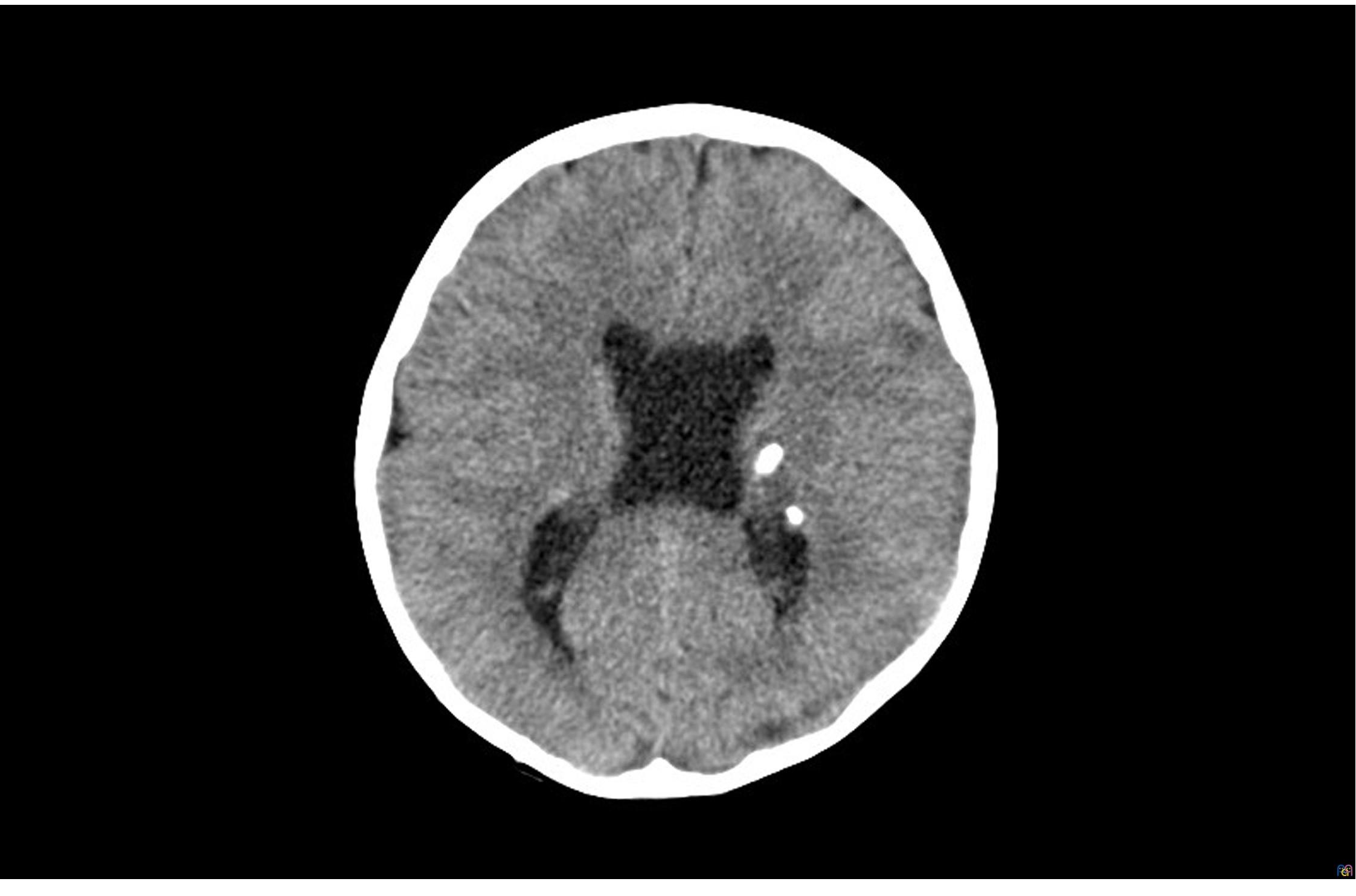
En Urgencias se realizan un análisis sanguíneo y de tóxicos en orina, con resultados normales. Se solicita una tomografía computarizada (TC) craneal (Fig. 1) en la que se identifican lesiones parenquimatosas, la mayoría calcificadas, con distribución periventricular.
| Figura 1. TC craneal (corte axial): lesiones parenquimatosas calcificadas periventriculares |
|---|
 |
Ante este cuadro se decide su ingreso para completar el estudio. En la planta de hospitalización se realiza un electroencefalograma (EEG) que muestra signos leves de afectación cerebral y anomalías epileptiformes intercríticas multifocales, registrándose una crisis eléctrica originada en región centro-temporal derecha sin manifestaciones clínicas. Dada la sospecha de esclerosis tuberosa, se amplía estudio cardiológico (sin hallazgos), renal (quistes bilaterales) y oftalmológico (hipermetropía), y se solicita estudio genético.
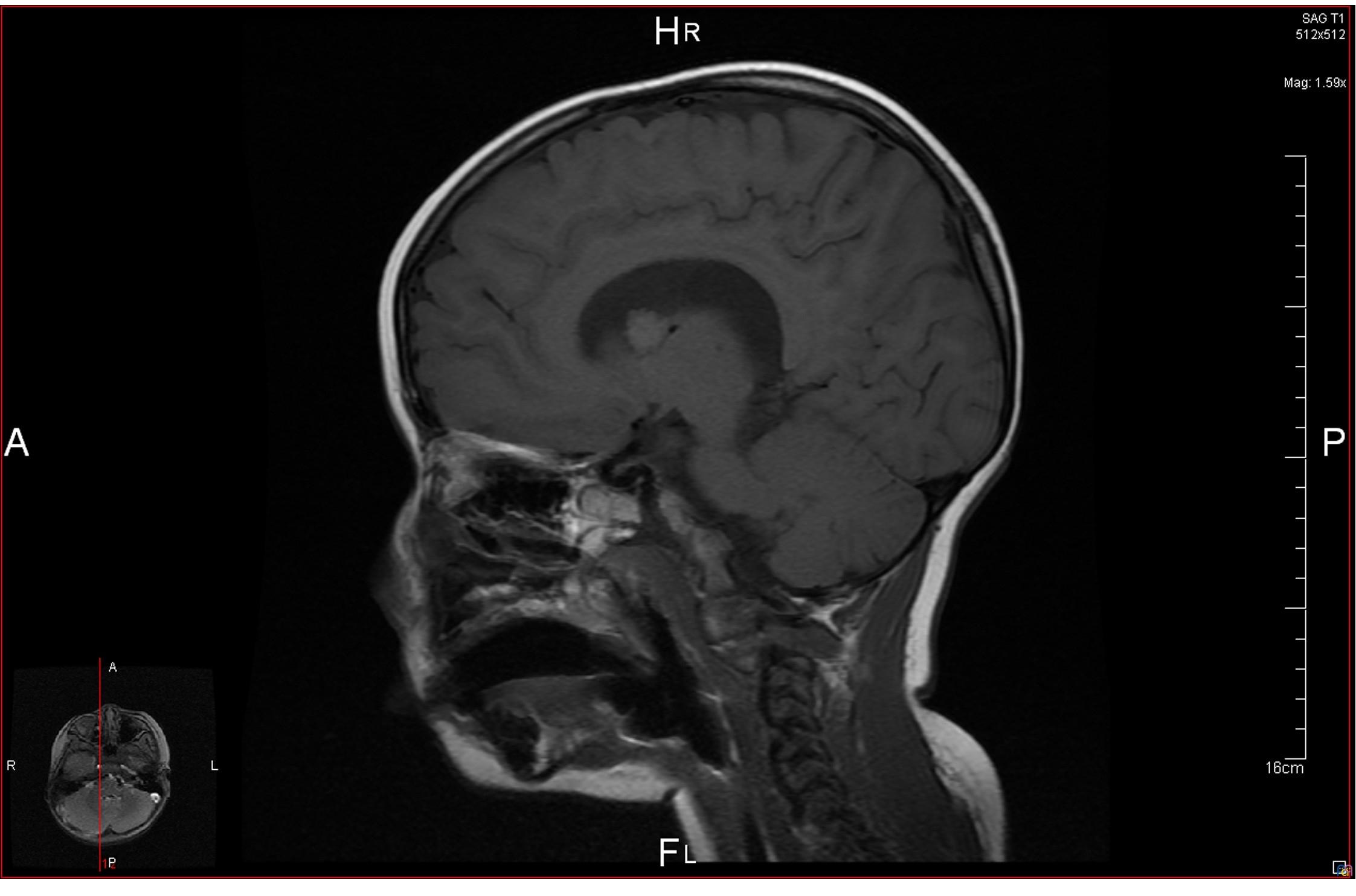
La resonancia magnética (RM) craneal (Fig. 2)muestra una imagen nodular de 11 mm cercana al agujero de Monro derecho, correspondiente a un astrocitoma subependimario de células gigantes (SEGA).
| Figura 2. RM craneal: imagen nodular de 11 mm de diámetro cercana al agujero de Monro |
|---|
 |
Al ingreso se inicia tratamiento antiepiléptico con ácido valproico (30 mg/kg/día), sin repetirse las crisis durante su estancia hospitalaria. Tras dos meses de seguimiento en consulta, refiere la persistencia de 1-2 crisis semanales, consistentes principalmente en desconexión del medio durante 15 segundos, con movimientos de cabeceo y mirada fija. Se asocia al tratamiento oxcarbazepina (30 mg/kg/día), con adecuado control posterior de las crisis.
Ha continuado seguimiento en consultas de Neurología (manteniendo tratamiento con ácido valproico y oxcarbazepina sin presentar nuevas crisis desde los cinco años), Nefrología (con imagen compatible con angiomiolipoma en último control ecográfico) y Dermatología (precisando sirolimus local como tratamiento de sus angiofibromas). El estudio genético demostró mutación en el gen TSC2.
DISCUSIÓN
La esclerosis tuberosa es un síndrome neurocutáneao caracterizado por el desarrollo de hamartomas en distintos órganos. Presenta una herencia autosómica dominante, aunque más del 60% de los casos se deben a mutaciones de novo. Es causado por mutaciones en los genes TSC1 y TSC2, que codifican para las proteínas hamartina y tuberina, respectivamente 1. La presentación clínica es muy variable, predominando las manifestaciones cutáneas, neurológicas, renales, cardiacas y oculares2.
Anomalías dermatológicas3,4:
- Máculas hipopigmentadas: presentes hasta en el 90% de los casos, principalmente en el tronco y las extremidades. Las formas pueden ser variables, siendo típicas en hoja de fresno y confeti.
- Angiofibromas: aparecen en el 75% de los casos como pápulas eritematosas centrofaciales.
- Hamartomas o nevus del tejido conectivo: pueden presentarse en forma de placa de Chagrin (65%), consistente en una placa rugosa sobreelevada, localizada fundamentalmente en región lumbosacra.
- Otras manifestaciones dermatológicas: fibromas periungueales y subungueales (Koenen), fibromas en mucosa oral, placas cutáneas en frente y cuero cabelludo.
Anomalías neurológicas:
- Crisis convulsivas: aparecen en el 80-90% de los pacientes y suele tratarse de la manifestación inicial. Frecuentemente se trata de crisis tónico-clónicas o espasmos infantiles5,6.
- Tumores cerebrales: la lesión cerebral característica es el tuber cortical (hamartomas de la sustancia gris localizados en las circunvoluciones de los hemisferios cerebrales y en la región subependimaria). Pueden calcificarse y protruir en la luz del ventrículo, lo que produce unas imágenes características en “goteo de vela”, u obstruir el agujero de Monro y provocar hipertensión intracraneal. Raramente evolucionan a astrocitomas malignos. El SEGA es característico del complejo esclerosis tuberosa. Todas estas lesiones pueden identificarse mediante TC craneal, especialmente si están calcificadas, siendo la RM más sensible7.
- Déficit cognitivo (en el 45-65% de los pacientes) y alteraciones del comportamiento (hiperactividad, inatención, autismo…)3,4.
Anomalías renales7:
- Angiomiolipomas (75-80%): tumores renales benignos, habitualmente pequeños, múltiples y bilaterales. Ocasionalmente pueden aparecer complicaciones por ruptura o sangrado. La incidencia aumenta con la edad.
- Quistes renales simples (20%): pueden llevar a hipertensión o insuficiencia renal.
- Carcinoma renal: menos frecuente.
Anomalías cardiacas7: los rabdomiomas cardiacos (50%) pueden detectarse en el periodo prenatal y suelen resolverse espontáneamente, aunque a veces causan arritmias o insuficiencia cardiaca. Se localizan preferentemente en los ventrículos y el septo.
Anomalías oculares: gliomas retinianos o del nervio óptico o nódulos elevados conocidos como lesiones en mora 4.
Otras: manifestaciones óseas (quistes, neoformación ósea perióstica, esclerosis), lesiones pulmonares (quistes, linfangioleiomiomatosis)7.
CONCLUSIÓN
La esclerosis tuberosa es una entidad infrecuente, con un amplio espectro de manifestaciones clínicas, incluyendo manifestaciones cutáneas, renales, neurológicas, cardiacas, oculares, óseas o pulmonares, dependiendo de la localización de los tumores. El diagnóstico se realiza predominantemente basándose en los hallazgos clínicos por lo que el conocimiento de esta entidad es fundamental en Atención Primaria 3. El manejo posterior requiere un abordaje multidisciplinar debido a la variabilidad clínica.
CONFLICTO DE INTERESES
Las autoras declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
EEG: electroencefalograma • RM: resonancia magnética • SEGA: astrocitoma subependimario de células gigantes • TC: tomografía computarizada.
BIBLIOGRAFÍA
- DiMario FJ, Sahin M, Ebrahimi-Fakhari D. Tuberous sclerosis complex. Pediatr Clin North Am. 2015;62:633-48.
- Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care. 2007;21:108-14.
- Rebolledo E, Cuen CD, Migoya A, Eudave LH. Esclerosis tuberosa: reporte de caso. Update J Med. 2013;2:3-6.
- Monteiro T, Garrido C, Pina R, Chorão R, Carrilho I, Figueiro S, et al. Esclerosis tuberosa: caracterización clínica e intento de correlación fenotipo/genotipo. An Pediatr (Barc). 2014;81:289-96.
- Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657.
- Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236-41.
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345-56.
- Yates JR, Maclean C, Higgins JN, Humphrey A, le Maréchal K, Clifford M, et al. The Tuberous Sclerosis 2000 Study: Presentation, initial assessmets and implicatios for diagnosis and management. Arch Dis Child. 2011;96:1020-5.