
Vol. 18 - Num. 72
Notas clínicas
¿En qué pensar ante una leucodermia y poliosis desde el nacimiento?
Júlia Morata Albaa, Andrés Tarragón Crosb
aServicio de Pediatría. Hospital Lluís Alcanyís. Játiva. Valencia. España.
bMIR-Pediatría. Hospital Lluís Alcanyís. Játiva. Valencia. España.
Correspondencia: J Morata. Correo electrónico: juliamorataalba@gmail.com
Cómo citar este artículo: Morata Alba J, Tarragón Cros A. ¿En qué pensar ante una leucodermia y poliosis desde el nacimiento? Rev Pediatr Aten Primaria. 2016;18:345-8.
Publicado en Internet: 10-10-2016 - Número de visitas: 31208
Resumen
El piebaldismo es una enfermedad hereditaria de la piel, autosómica dominante, por mutación del gen KIT, que cursa con lesiones de piel con hipopigmentación (leucodermia) y mechón de pelo blanco (poliosis) desde el nacimiento. Las lesiones son permanentes y sin progresión.
Palabras clave
● Hipopigmentación ● Mechón blanco ● PiebaldismoINTRODUCCIÓN
El piebaldismo es una enfermedad hereditaria de la piel con herencia autosómica dominante. Afecta por igual a ambos sexos y sin predominio de razas. Su incidencia es de menos de uno por cada 20 000 personas1.
Se caracteriza por presentar desde el nacimiento un mechón de pelo blanco (poliosis) y áreas cutáneas despigmentadas (leucodermia) por ausencia congénita de melanocitos en las zonas afectadas por mutación del gen KIT2. Dicha mutación afecta a la diferenciación y migración de los melanoblastos, por lo que el curso de la enfermedad es permanente y estático desde el nacimiento1.
La clínica y gravedad del fenotipo dependen del sitio de la mutación en el gen KIT, siendo la forma clásica el piebaldismo por mutación en las inmediaciones del codón 620.
Es relevante destacar que, salvo las lesiones cutáneas y del pelo, los pacientes están sanos. Esto es importante para realizar el diagnóstico diferencial con otras enfermedades.
CASO CLÍNICO
Niña de un mes y medio de edad que es remitida a la consulta de Pediatría por un riñón derecho pélvico y un mechón blanco de pelo en la región frontal (Figs. 1 y 2).
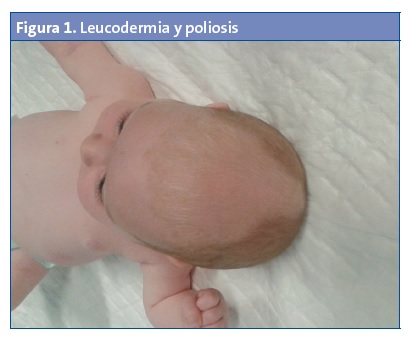
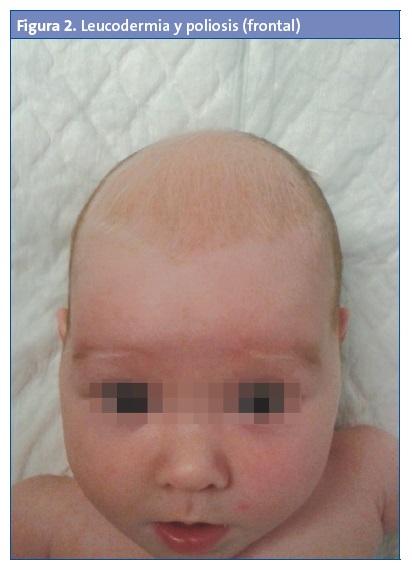
Procede de la primera gestación de una madre sana. Embarazo controlado sin incidencias, con ecografías normales salvo por el riñón derecho pélvico. Parto vaginal, instrumentado a la edad gestacional de 38 semanas. Peso al nacimiento de 2320 gramos, Apgar 10/10. Otoemisiones correctas bilaterales.
Antecedentes familiares: padres de raza blanca, madre sana de 25 años, padre sano de 36 años, salvo manchas acrómicas en el tercio medio de ambos muslos, sin haber sido diagnosticado de ninguna enfermedad. Se desconocen otros antecedentes familiares.
La paciente está asintomática; su ritmo intestinal y diuresis son normales. En la exploración física presenta un mechón de pelo blanco frontal y una mácula acrómica de forma triangular en la región frontal. No tiene otras lesiones cutáneas. El resto de la exploración es normal, con desarrollo psicomotor hasta el momento normal.
Se sospecha clínicamente un piebaldismo, pero se solicitan pruebas complementarias para comprobar que no presenta anomalías oftalmológicas ni sordera, que deben estar ausentes para confirmar el diagnóstico clínico de piebaldismo. Con la valoración oftalmológica se descartaron lesiones oculares y se realizaron potenciales evocados auditivos, que fueron normales.
En la ecografía abdominal se confirmó la presencia de un riñón derecho pélvico sin otras anomalías.
Se solicitó estudio analítico (hemograma y química), que fue normal, y estudio genético para mutación del gen KIT (OMIM 164920) (4q12), que fue negativo. Se detectó una variante en heterocigosis en el gen KIT de significado clínico desconocido [c.2509G>A;(p.Ala837Thr)] (en el padre también resultó negativo).
DISCUSIÓN
En la leucodermia o disminución de la pigmentación de la piel se deben diferenciar dos causas: la ausencia de melanocitos, como en el piebaldismo, y la presencia normal de melanocitos, pero con poca melanina, como en el albinismo.
El diagnóstico del piebaldismo es clínico. Las únicas alteraciones son las máculas cutáneas despigmentadas y el mechón de pelo blanco. Las lesiones cutáneas suelen ser simétricas. El 90% de los casos presentan el mechón blanco de pelo y el área acrómica en región frontal, que puede extenderse a la parte media de las cejas y pestañas. Otras zonas de lesiones características son: desde la mitad de los brazos hasta las muñecas, desde la mitad de los muslos hasta la mitad de las piernas, tórax o abdomen. En ocasiones se objetivan máculas hiperpigmentadas en el interior de las máculas acrómicas3,4.
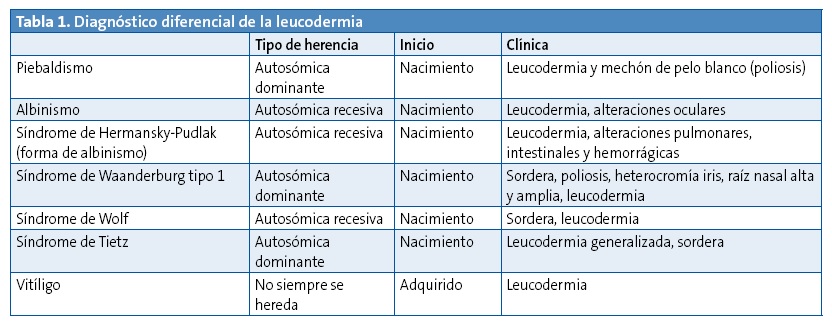
El diagnóstico diferencial se debe hacer con otras entidades que cursan con falta de pigmentación de la piel (Tabla 1), de las que se diferencia porque aquellas presentan otra sintomatología, además de la cutánea.
El piebaldismo está relacionado con una mutación en el gen KIT. Este gen codifica una proteína tirosina quinasa, que es esencial para la proliferación y la migración de las células de la cresta neural, precursoras de la célula germinal y de las células sanguíneas. El gen KIT también es importante en la diferenciación y migración de los melanoblastos. La mutación identificada en el gen KIT para el piebaldismo tiene su locus en el cromosoma 4q12. Este gen codifica el receptor de membrana de la tirosina y para un factor de crecimiento embrionario llamado mast cell growth factor, stem cell growth factor o steel factor2. Se han descrito más de 40 mutaciones diferentes en este gen (heterogeneidad alélica) y los fenotipos más graves se asocian cuando la mutación en el exón 2 del gen KIT involucra el dominio intracelular tirosina quinasa, produciéndose un cambio del aminoácido cisteína por glicina, y por tanto, un trastorno en la polaridad de la proteína; mientras que fenotipos más leves se han correlacionado con mutaciones en el dominio extracelular amino-terminal2.
A pesar de que el piebaldismo es una enfermedad benigna y estática, es importante el seguimiento de los pacientes por el problema psicosocial que pueden presentar a lo largo de los años. A nivel dermatológico, existen algunos tratamientos médicos para intentar corregir la falta de pigmentación de la piel y en ocasiones se recurre a microinjertos de melanocitos autólogos. No precisa otros tratamientos pues no existen más anomalías que tratar.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
BIBLIOGRAFÍA
- Cabrera NB, Breto AG, Castro M, Torres MR, Milián RI. Piebaldismo en un recién nacido. Leucodermia rara. Rev Ciencias Médicas. 2013;17:92-9.
- Bondanza S, Bellini M, Roversi G, Raskovic D, Maurelli R, Palonni E, et al. Piebald trait: implication of KIT mutation on in vitro melanocyte survival and on the clinical application of cultured epidermal autografts. J Invest Dermatol. 2007;127:676-86.
- Valdivielso-Ramos M, Mauleón C, Martín MA, Balbín E, De la Cueva P, Hernanz JM. Piebaldismo. Acta Pediatr Esp. 2011;69:464-5.
- Perelló-Alzamora MR, Alonso-San Pablo MT, Unamuno P. Mechón blanco frontal aislado. Diagnóstico y comentario. Piel. 2011;26:515-6.