
Vol. 17 - Num. 68
Notas clínicas
Debilidad muscular recurrente en las piernas. No pensar siempre en molestias de crecimiento
Júlia Morata Albaa, M.ª Isabel Jiménez Candelb
aServicio de Pediatría. Hospital Lluís Alcanyís. Játiva. Valencia. España.
bServicio de Pediatría. Hospital Virgen del Castillo. Yecla. Murcia. España.
Correspondencia: J Morata. Correo electrónico: juliamorataalba@gmail.com
Cómo citar este artículo: Morata Alba J, Jiménez Candel MI. Debilidad muscular recurrente en las piernas. No pensar siempre en molestias de crecimiento. Rev Pediatr Aten Primaria. 2015;17:341-5.
Publicado en Internet: 27-10-2015 - Número de visitas: 31057
Resumen
La parálisis periódica hiperpotasémica es una canalopatía del músculo esquelético que se caracteriza por episodios recurrentes de debilidad muscular que pueden ser desencadenados por el ejercicio, el frío, el reposo o poco después del ejercicio y por aporte de potasio.
Describimos el caso de una niña de cuatro años de edad, con antecedentes familiares de parálisis periódica hiperpotasémica y con algún episodio de debilidad en miembros inferiores. Asintomática hasta unos seis meses antes de acudir. Refieren unos tres episodios de debilidad muscular y sensación de adormecimiento de miembros inferiores, por las noches, que se resolvieron espontáneamente en unos minutos. No presenta otra clínica asociada.
Palabras clave
● Debilidad muscular recurrente ● Parálisis periódicaINTRODUCCIÓN
Las parálisis periódicas son un trastorno poco frecuente (prevalencia de 1 caso/200 000 habitantes), que cursan con episodios de debilidad muscular aguda que puede confundirse con otras enfermedades como la epilepsia o la miastenia gravis, entre otras. Mujeres y hombres parecen afectados por igual.
La parálisis periódica hiperpotasémica se incluye dentro de las canalopatías musculares. En esta entidad se producen mutaciones en el gen SCN4A, que codifica la subunidad alfa del canal del sodio del músculo esquelético, que se localiza en el cromosoma 17q23-251. El patrón de herencia es autosómico dominante2-4 y es una entidad poco frecuente2.
Los defectos estructurales o funcionales en el canal del sodio del músculo esquelético causados por mutaciones en el gen SCN4A consisten, por lo general, en sustituciones de aminoácidos que impiden la inactivación del canal que sigue normalmente a un potencial de acción, lo que ocasiona un flujo incontrolado de sodio hacia el interior de la fibra muscular. Como consecuencia, la fibra se despolariza y ello impide la generación de nuevos potenciales de acción. La entrada masiva de sodio dentro de las células provoca una salida de potasio que explica sus concentraciones elevadas en la sangre2,4.
Se caracteriza por ataques recurrentes de debilidad muscular que pueden durar de minutos a horas y se asocian por lo general, pero no siempre, a un aumento de las concentraciones del potasio sérico (en algunas series los niveles de potasio sérico registrados durante los ataques de debilidad muscular fueron de 5,3 mEq/l) y de la creatinfosfocinasa5. Por lo general, no se afectan los músculos respiratorios y después de un ataque puede persistir debilidad muscular moderada durante uno o dos días. Los síntomas suelen aparecer de modo temprano, en la primera década de la vida.
Se han descrito tres variantes de parálisis periódica hiperpotasémica: la forma pura sin otros síntomas añadidos, la forma que evoluciona con miotonía clínica o electromiográfica, y la que evoluciona con paramiotonía2.
CASO CLÍNICO
Niña de tres años y diez meses de edad que es valorada en consultas por episodios recurrentes de debilidad muscular en miembros inferiores, con recuperación espontánea en minutos. Antecedentes personales sin interés. Embarazo controlado sin incidencias; nacimiento por parto vaginal. Lactancia artificial. Desarrollo perinatal y psicomotor normal. Ni debilidad muscular ni calambres referidos hasta seis meses antes.
En los antecedentes familiares destaca parálisis hiperpotasémica familiar en varios familiares de la rama paterna (padre, tío y tía paterna, abuela paterna) sin aportar más datos y sin saber precisar estudio genético.
La clínica se inició unos seis meses antes de acudir a la consulta, estando previamente asintomática; refirieron 3-4 episodios intermitentes, en días separados entre sí, de calambres musculares y sensación de adormecimiento de miembros inferiores mientras dormía, de los que se recuperó espontáneamente en unos minutos. No muestra otra clínica, ni afectación de otros miembros. Desde estos episodios la paciente permaneció asintomática.
Exploración clínica y neurológica normal en el momento de ser valorada en consulta, con fuerza, sensibilidad y reflejos osteotendinosos normales.
Ante la sospecha de parálisis hiperpotasémica familiar, se solicitó estudio analítico con hemograma y bioquímica, siendo normales. Hemograma normal, electrolitos en sangre y orina normales, K 4,5 mEq/l con excreción fraccional de potasio (EFK) del 13%. No fue posible realizar análisis de sangre coincidiendo con la clínica.
Tampoco se pudo realizar coincidiendo con la clínica la electromiografía (EMG), la determinación de creatinfosfocinasa (CPK) y el electrocardiograma (ECG), pues no presentó de nuevo síntomas. La electromiografía (EMG) sin coincidir con la clínica fue normal.
Ante la sospecha clínica, dado los antecedentes familiares, a pesar de que la clínica no era muy manifiesta, se solicitó estudio genético del gen SCN4A (OMIM +603967), detectando mutación en heterocigosis p.Thr704Met.
En el momento del diagnóstico no se inició tratamiento con acetazolamida por vía oral dado que los episodios de debilidad muscular no se han repetido y los valores de potasio sérico fueron normales en los controles realizados. Se realizarán controles clínicos y analíticos evolutivos periódicos para valorar si, en el algún momento, precisa iniciar tratamiento médico. Se pautó dieta no rica en potasio.
DISCUSIÓN
En la parálisis periódica hiperpotasémica los episodios de debilidad muscular ocurren por lo general por la mañana, al despertar, y al final del día6. Como factores desencadenantes se señalan el ejercicio, el frío, el reposo poco después del ejercicio y el aporte de potasio6.
Durante los ataques, los reflejos osteotendinosos están disminuidos o ausentes, las concentraciones de potasio sérico se elevan y puede observarse un incremento de la amplitud de las ondas T en el ECG, un QT largo o intervalo QU largo sugestivo de síndrome de Andersen. En nuestro caso no se pudo realizar la exploración clínica ni la determinación analítica ni el ECG coincidiendo con episodios de debilidad muscular.
Entre los episodios, los niños pueden presentar miotonía en los párpados, cara, cuello, lengua e incluso en las extremidades6, o estar asintomáticos.
En los pacientes con parálisis periódica hiperpotasémica puede no observarse clínicamente el fenómeno miotónico, pero sí evidenciarse en la electromiografía7.
En nuestra paciente no se observó fenómeno miotónico clínico ni al realizar la electromiografía, pero no se pudo realizar el estudio durante un episodio de debilidad muscular.
La parálisis periódica hiperpotasémica puede ser difícil de diagnosticar cuando los pacientes son evaluados después o entre los ataques, cuando la fuerza del músculo y los niveles séricos de potasio son normales. La creatinfosfocinasa está frecuentemente elevada entre los ataques de parálisis, pero esto es poco sensible y nada específico de la entidad.
Según Narberhaus et al.2, en la parálisis periódica hiperpotasémica el estudio histológico del músculo no aporta información de interés, aunque en la microscopía óptica pueden observarse hallazgos compatibles con miopatía vacuolar, mientras que en la electrónica pueden detectarse túbulos T dilatados6.
En el tratamiento preventivo de los episodios de debilidad muscular se recomienda el empleo de inhibidores de la anhidrasa carbónica, como la acetazolamida, dos o tres veces en el día, porque disminuyen la frecuencia de los ataques y puede mejorar la miotonía si está presente. También se sugiere como tratamiento preventivo el uso continuo de diuréticos como la hidroclorotiazida y el empleo de agonistas β-adrenérgicos como el albuterol o salbutamol, y evitar el frío5 y alimentos ricos en potasio8.
Para el tratamiento de episodios agudos, graves y prolongados se sugiere la administración de gluconato de calcio por vía intravenosa, glucosa intravenosa o glucosa e insulina intravenosas e hidroclorotiazida vía intravenosa8.
La mayoría de las parálisis periódicas hiperpotasémicas se observan en varios miembros de una familia y se transmiten de forma autosómica dominante, como en nuestro caso. Los familiares directos tienen una probabilidad del 50% de presentar la mutación4. Están descritas varias mutaciones en el gen SCN4A asociadas a la enfermedad.
Existen casos de presentación esporádica y se han descrito muy raramente formas secundarias de parálisis hiperpotasémica. Puede ocurrir hiperpotasemia debido a otras causas, como por ejemplo en pacientes con hemorragia intestinal o estados hipercatabólicos, en la hemólisis intravascular, transfusiones de sangre incompatibles, quemaduras, tratamiento quimioterapéutico de tumores sólidos o leucemia, en la insuficiencia renal aguda y crónica, hiperplasia suprarrenal congénita, anemia drepanocítica, lupus eritematoso sistémico, uropatías obstructivas, periodo postrasplante renal, amiloidosis renal, durante el empleo de diuréticos como la espironolactona, en la enfermedad de Addison, entre otras9,10.
CONCLUSIONES
Ante todo paciente con debilidad muscular aguda recurrente debe realizarse una anamnesis y exploración clínica completa para una correcta orientación diagnóstica.
Se debe realizar el diagnóstico diferencial de otras parálisis periódicas, así como de otras causas de cuadriparesia aguda, incluyendo síndrome de Guillain-Barré, botulismo, y mielopatía cervical. Estas entidades se diferencian de la parálisis hiperpotasémica familiar periódica por los valores elevados de potasio en esta última, así como por la ausencia de afectación respiratoria y recuperación espontánea en unas horas (Tabla 1).
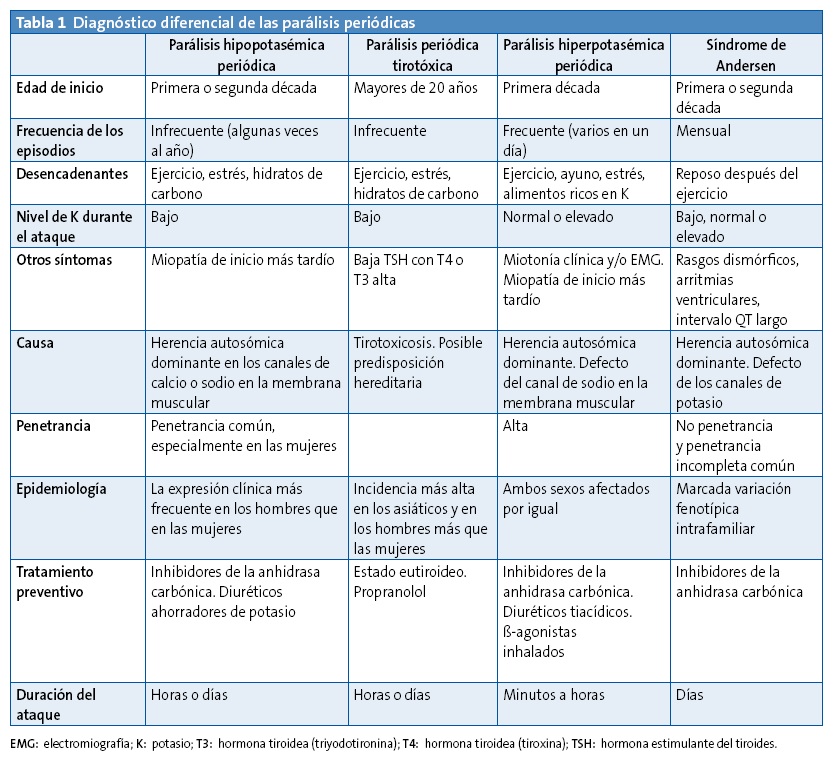
A pesar de ser una entidad poco frecuente, no debemos olvidar la posibilidad de una parálisis periódica hiperpotasémica, así como también la posibilidad de parálisis periódicas hipopotasémicas y normopotasémica; estas últimas consideradas variantes de la parálisis periódica hiperpotasémica. El diagnóstico de esta enfermedad se sospecha por la clínica y se puede confirmar con la determinación de electrolitos en sangre y con confirmación genética.
En ocasiones la sintomatología puede manifestarse por la noche, como en nuestro caso, tras el reposo después del ejercicio diario habitual de un niño, y debemos pensar que no siempre esta sintomatología es secundaria a los dolores de crecimiento.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS: CPK: creatinfosfocinasa • ECG: electrocardiograma • EMG: electromiografía • EFK: excreción fraccional de potasio.
BIBLIOGRAFÍA
- Lee SC, Kim HS, Park YE, Choi YC, Park KH, Kim DS. Clinical diversity of SCN4A-mutation-associated skeletal muscle sodium channelopathy. J Clin Neurol. 2009;5:186-9.
- Narberhaus B, Cormand B, Cuenca E, Ribasés M, Monells J. Parálisis periódica hipercaliémica: presentación de una familia española con la mutación p.Thr 704 Met en el gen SCN4A. Neurología. 2008;23:427-35.
- Lossin C, Nam TS, Shahangian S. Altered fast and slow inactivation of the N440K Nav1.4 mutant in a periodic paralysis syndrome. Neurology. 2012;79:1033.
- Vicart S, Sternberg D, Fournier E. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology. 2004;63:2120.
- Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3-23.
- Ruggieri VL, Arberas CL. Canalopatías hereditarias neuromusculares: miotonías no distróficas, paramiotonías y parálisis periódicas. Rev Neurol. 2002;34:150-6.
- Fournier E, Arzel M, Sternberg D. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. 2004;56:650.
- Venance SL, Cannon SC, Fialho D. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129:8.
- Mena V. Alteraciones electrolíticas. En: De la Torre E, Pelayo EJ (eds.). Pediatría. La Habana: Editorial de Ciencias Médicas; 2006. p. 558-85.
- Clausen T. Hormonal and pharmacological modification of plasma potassium homeostasis. Fundam Clin Pharmacol. 2010;24:595-605.