

Síndrome de Gitelman, a propósito de un caso
Júlia Morata Albaa, A Hervás Andrésb
aServicio de Pediatría. Hospital Lluís Alcanyís. Játiva. Valencia. España.
bServicio de Pediatría. Hospital Lluís Alcanyís. Xàtiva. Valencia. España.
Correspondencia: J Morata. Correo electrónico: juliamorataalba@gmail.com
Cómo citar este artículo: Morata Alba J, Hervás Andrés A. Síndrome de Gitelman, a propósito de un caso. Rev Pediatr Aten Primaria. 2015;17:e183-e187.
Publicado en Internet: 10-09-2015 - Número de visitas: 23145
Resumen
Las tubulopatías son un grupo heterogéneo de entidades definidas por anomalías de la función tubular renal. El síndrome de Gitelman, objeto de nuestro caso, está causado por mutaciones inactivantes del gen SLC12A3, que codifica el cotransportador Na-Cl sensible a tiazidas del túbulo contorneado distal, produciendo así una pérdida urinaria de Cl-Na. Suele iniciarse al final de la infancia o en la adolescencia, con avidez por las comidas saladas, cansancio excesivo, debilidad y calambres musculares. Una pequeña proporción de niños puede tener fallo de medro. Característicamente, no presentan hipertensión arterial ni disminución de la función glomerular. En algunos pacientes las manifestaciones clínicas son leves y pueden pasar desapercibidas.
Palabras clave
● Potasio ● Síndrome de GitelmanINTRODUCCIÓN
Las tubulopatías son un grupo heterogéneo de entidades definidas por anomalías de la función tubular renal. Se distinguen las tubulopatías hereditarias o primarias de las secundarias a tóxicos, fármacos u otras enfermedades. Pueden ser simples o complejas, según se afecte el transporte tubular de una o varias sustancias.
Dependiendo de la función tubular que se encuentre afectada, cada entidad tiene una edad de aparición, manifestaciones clínicas y analíticas, gravedad y pronóstico propios. Se pueden clasificar según la zona del túbulo afectada o según su mecanismo fisiopatológico.
El diagnóstico de sospecha de las distintas tubulopatías se basa en:
- Anamnesis: historia familiar (consanguinidad), antecedentes obstétricos y neonatales, síntomas sugestivos, historia dietética, avidez por la sal, ingesta de agua y diuresis.
- Exploración: crecimiento y desarrollo (peso, talla, presión arterial), exploración general, hidratación, raquitismo, anomalías en órganos de los sentidos.
- Evaluación de la función renal:
- Orina: inspección, tiras reactivas y sedimento.
- Volumen urinario (orina de 24 horas o corregido por 100 ml de filtrado glomerular).
- Cálculo del filtrado glomerular renal.
- Función renal (excreciones fraccionales de bicarbonato Na, K, Cl, ácido úrico, tasa de reabsorción tubular de fosfato).
- Excreción de Ca, Mg, glucosa, proteínas de bajo peso molecular (calculada en orina de 24 horas, o cocientes urinarios).
- Otros estudios: ecografía renal, estudios oftalmológicos, de audición y estudio genético.
Presentamos un caso de una niña afecta de síndrome de Gitelman. Esta tubulopatía está causada por mutaciones inactivantes del gen SLC12A3 (OMIM #263800), que codifica el cotransportador Na-Cl sensible a tiazidas del túbulo contorneado distal, produciendo una pérdida urinaria de Cl-Na con la depleción moderada de volumen secundaria y estimulación del sistema renina-angiotensina-aldosterona, con hipopotasemia y alcalosis metabólica secundaria. Se evidencia una reducción en la expresión del canal de Mg TRPM6 en la membrana apical de las células del túbulo contorneado distal que es responsable de la hipomagnesemia y un aumento en la reabsorción de calcio que causa la hipocalciuria.
Muchos pacientes se encuentran asintomáticos o presentan síntomas leves neuromusculares de inicio en la infancia tardía o en la juventud; no obstante, existen formas más graves en la primera infancia, difíciles de distinguir clínicamente de un síndrome de Bartter tipo III. No existe correlación fenotipo-genotipo y los síntomas principales son avidez por alimentos salados, presión arterial normal o baja, intolerancia al ejercicio físico, retraso de crecimiento y talla baja. Los síntomas neuromusculares son leves, secundarios a hipopotasemia e hipomagnesemia, pero puede haber tetania o convulsiones. Puede existir prolongación del QT y arritmias.
El tratamiento se basa en la administración de cloruro potásico y sales de magnesio (en forma de cloruro, lactato o aspartato). En situaciones graves se administran por vía intravenosa. Se pueden asociar diuréticos ahorradores de potasio o antagonistas de la aldosterona. Se deben evitar los ejercicios físicos intensos para minimizar las arritmias, junto a una dieta rica en potasio, magnesio y sodio. No se deben tomar fármacos arritmogénicos.
CASO CLÍNICO
Niña de tres años de edad que se diagnostica de neumonía con condensación parenquimatosa en lóbulo inferior derecho.
En los antecedentes personales no hay datos de interés. Embarazo controlado sin incidencias, nacida a las 38 semanas de edad gestacional, cesárea por falta de progresión. Peso al nacimiento de 2870 g. Vacunas y desarrollo psicomotor normal. Refieren desde el primer año de vida que bebe bastantes líquidos, con diuresis abundante. Peso y talla en el percentil 50. Refieren avidez por la sal (se bebe el agua de la playa con avidez y prefiere alimentos salados) y calambres musculares en ocasiones. No hay antecedentes familiares a destacar.
Peso 12,4 kg (p10-25), talla 95 cm (p50), tensión arterial 100/60 mmHg, frecuencia cardiaca 90 lpm, frecuencia respiratoria 28 rpm, temperatura 38,3 °C. Regular aspecto general por facies febril, decaída y discreta sequedad de mucosas. Cráneo normal. Signos meníngeos negativos. No presenta exantemas ni petequias. Otoscopia y orofaringe normal. A la auscultación se observa una disminución de la entrada de aire en base derecha. Abdomen blando y depresible, no se palpan masas, ni visceromegalias. Genitales normales de niña. Región lumbosacra normal. Resto de la exploración normal.
En los análisis realizados para valorar parámetros de infección se objetivó leucocitosis de 22 000 con neutrofilia (neutrófilos 87%) y proteína C reactiva (PCR) de 200 mg/l, pero destacaba la hipopotasemia de 2,2 mmol/l con Na 129 mmol/l, Cl 92,2 mmol/l, Ca 9,5 mg/dl, Mg 1,7 mg/dl, Cr 0,5 mg/dl y alcalosis metabólica (pH 7,50 y bicarbonato 24,1 mmol/l). Renina y aldosterona elevadas.
Se solicitó estudio de orina: densidad 1005, PH 8, sedimento normal. Iones en orina: K 17,5 mEq/l, Cl 10 mEq/l, Na 7 mEq/l; Na orina/K orina 0,4. Excreciones fraccionadas de K elevadas (8,32%), de Na (0,06%) y de Cl (0,11%) en valores de normalidad. Ca/Cr 0,56 mg/mg. Aclaramiento de creatinina normal, filtrado glomerular 104,50 ml/min/1,73m2,
La ecografía renal fue normal y en el electrocardiograma (ECG) se confirmó la hipopotasemia por prolongación del PR en el ECG.
Ante el diagnóstico de neumonía basal derecha, se inició tratamiento antibiótico con cefuroxima intravenosa y de modo progresivo fue mejorando clínica, radiológica y analíticamente.
Ante la sospecha de tubulopatía, dada la hipopotasemia y la pérdida urinaria elevada de este electrolito, se inició tratamiento con suplementos intravenosos de potasio y se corrigieron poco a poco las cifras, siendo al alta el K de 3,9 mmol/l.
Se controló en consultas de Nefrología Pediátrica, confirmándose la tubulopatía genéticamente (síndrome de Gitelman) y manteniendo adecuados niveles de K sérico con suplementos orales y dieta rica en este electrolito.
La paciente es portadora de las variaciones p.Gly741Arg y p.Cys994Tyr, ambas en heterocigosis, en el gen SLC12A3, localizado en el brazo largo del cromosoma 16. Transmisión autosómica recesiva.
Nuestra paciente, desde su diagnóstico, recibe una dieta rica en potasio y magnesio y suplementos orales de potasio. Tras varios años de tratamiento, dada las altas dosis de suplementos orales de K (4,3 mEq/kg/día) para mantener niveles séricos de K 3,1-3,5 mmol/l, se decide asociar al tratamiento diuréticos ahorradores de potasio.
En ocasiones, coincidiendo con episodios de diarrea y vómitos, presenta hipopotasemia sintomática, que precisa suplementos intravenosos de potasio.
No hay datos de hipomagnesemia en ningún momento, por lo que no recibe suplementos de magnesio orales, solo una dieta rica en magnesio.
Se trata de una enfermedad de carácter permanente con evolución y pronóstico favorables si se cumple el tratamiento.
DISCUSIÓN
Es importante conocer la existencia de las tubulopatías para poder pensar en ellas en el momento de hacer un diagnóstico. Son entidades poco frecuentes, pero lo suficientemente importantes como para realizar un correcto diagnóstico.
En nuestra paciente el diagnóstico se sospechó ante el hallazgo de hipopotasemia coincidiendo con el diagnóstico de una neumonía que precisó ingreso hospitalario.
Ante la hipopotasemia se sospechó una tubulopatía. En el momento del diagnóstico los valores de excreción urinaria de K y Mg no eran muy elevados, pero la anamnesis completa nos reafirmaba la sospecha diagnóstica, pues la paciente había presentado desde hacía varios años avidez por la sal, cansancio y calambres musculares. Se inició tratamiento con suplementos de potasio intravenosos ante el riesgo de arritmias.
Tras finalizar el tratamiento antibiótico por la neumonía y tras la normalización de las cifras de potasio sérico, la paciente fue dada de alta con suplementos orales de potasio y ha seguido controles periódicos de los electrolitos en sangre y orina.
En el síndrome de Gitelman, los datos analíticos típicos son la alcalosis metabólica, hipopotasemia e hipomagnesemia, con pérdida renal excesiva de potasio y magnesio e hipocalciuria. En ocasiones la hipomagnesemia no es objetivada hasta varios años después del diagnóstico.
Es importante comunicar a los pacientes que sigan una dieta rica en potasio y magnesio y que eviten medicamentos que alarguen el QT por el riesgo de arritmias que estos podrían ocasionar.
Un hecho a tener en cuenta es que en procesos de diarrea puede existir una pérdida elevada de potasio, que ocasionaría un empeoramiento de los síntomas por descenso de los valores de potasio sérico, que pueden precisar aportes intravenosos de este electrolito.
CONCLUSIONES
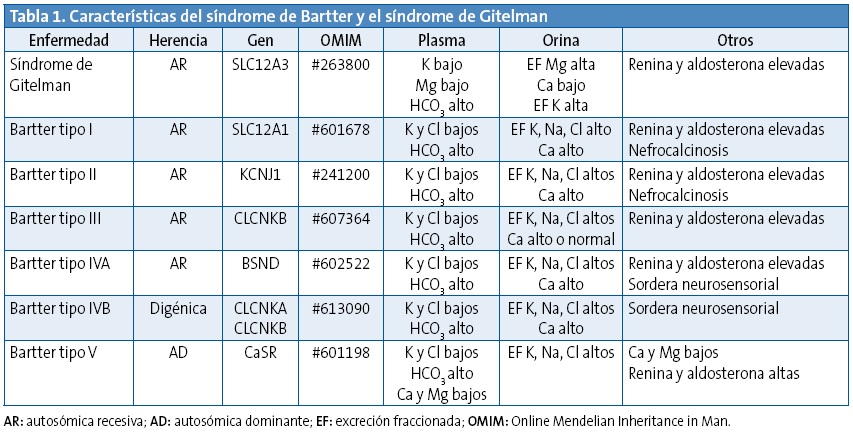
Las tubulopatías se pueden presentar en un contexto clínico diferente, pero debemos pensar en algunas de ellas (como el síndrome de Bartter o Gitelman) (Tabla 1) ante síntomas de cansancio, calambres musculares o avidez por la sal. Una primera aproximación diagnóstica, ante la sospecha clínica, se puede realizar con un análisis de sangre con electrolitos y con el cálculo de pérdidas urinarias de algunos electrolitos en la primera orina de la mañana. En el caso del síndrome de Gitelman, una vez confirmada la alteración electrolítica en sangre (hipopotasemia con o sin hipomagnesemia) y orina (pérdida urinaria excesiva de K y Mg con hipocalciuria) se debe remitir a Nefrología Pediátrica para realizar el estudio genético e iniciar el tratamiento con suplementos orales de K y/o Mg y realizar el seguimiento oportuno.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS: ECG: electrocardiograma • PCR: proteína C reactiva.
BIBLIOGRAFÍA
- Ariceta G, Rodríguez Soriano J. Inherited tubulopathies with metabolic alkalosis. Semin Nephrol. 2006;26:422-33.
- Asociación Española de Nefrología Pediátrica. Nefrología Pediátrica. Manual práctico. Madrid: Editorial Médica Panamericana; 2011.
- Viganò C, Amoruso C, Barretta F, Minnici G, Albisetti W, Syrèn ML, et al. Renal phosphate handling in Gitelman syndrome, the results of a case-control study. Pediatr Nephrol. 2013;28:65-70.
- Scholl UI, Dave HB, Lu M, Farhi A, Nelson-Williams C, Listman JA, et al. SeSAME/EAST syndrome–phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol. 2012;27:2081-90.
- Sinha A, Lněnička P, Basu B, Gulati A, Hari P, Bagga A. Gitelman syndrome: novel mutation and long-term follow-up. Clin Exp Nephrol. 2012;16:306-9.
- Tammaro F, Bettinelli A, Cattarelli D, Cavazza A, Colombo C, Syrén ML, et al. Early appearance of hypokalemia in Gitelman syndrome. Pediatr Nephrol. 2010;25:2179-82.
- Landau D. Potassium handling in health and disease: lessons from inherited tubulopathies. Pediatr Endocrinol Rev. 2004;2:203-8.
- Chan CF, Mu SC, Lau BH, Chang CJ, Lin SH. Gitelman’s syndrome: report of one case. Acta Paediatr Taiwan. 2008;49:31-4.