
Vol. 27 - Num. 105
Notas clínicas
Histiocitosis de células de Langerhans. Inicio con exantema vesicular
María Aldana Villamañána, Ignacio Aldana Villamañánb, Javier Aldana Gómezc
aMIR-Pediatría. Hospital Universitario de Torrejón. Torrejón de Ardoz. Madrid. España.
bPediatra. CS Segovia 3. Segovia. España
cPediatra. CS Segovia I. Segovia. España.
Correspondencia: J Aldana. Correo electrónico: aldana.0001@gmail.com
Cómo citar este artículo: Aldana Villamañán M, Aldana Villamañán I, Aldana Gómez J. Histiocitosis de células de Langerhans. Inicio con exantema vesicular . Rev Pediatr Aten Primaria. 2025;27:85-8. https://doi.org/10.60147/9c9744d1
Publicado en Internet: 11-03-2025 - Número de visitas: 7030
Resumen
La histiocitosis de células de Langerhans constituye un grupo de enfermedades poco frecuentes caracterizadas por la infiltración tumoral de casi cualquier órgano del cuerpo por células del sistema fagocítico mononuclear. La afectación ósea es la más frecuente, seguida de las lesiones cutáneas. Otros órganos implicados son los ganglios linfáticos, la médula ósea, el bazo, el hígado, el pulmón, la glándula pituitaria y el sistema nervioso central. La gran variedad de manifestaciones clínicas cuando el debut es cutáneo, simulando otras enfermedades más frecuentes, a menudo provoca un retraso en el diagnóstico que condiciona un peor pronóstico de supervivencia y de secuelas posteriores. Un alto índice de sospecha ante lesiones cutáneas de variada índole o de presentación atípica llevarán a un diagnóstico precoz de histiocitosis.
Palabras clave
● Células de Langerhans ● Exantema vesicular ● HistiocitosisCASO CLÍNICO
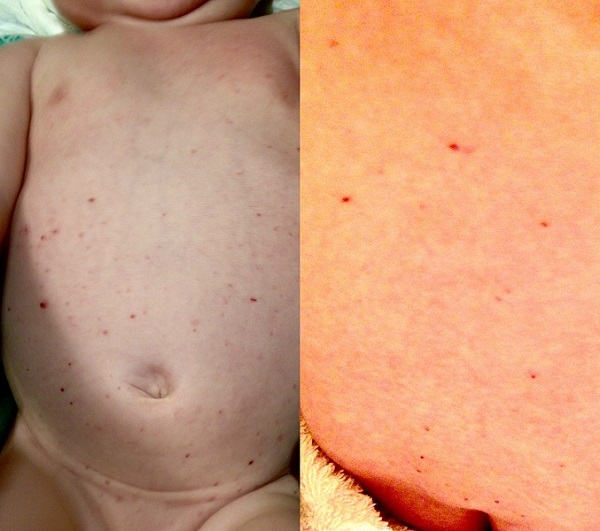
Lactante de sexo femenino y 3 meses de edad con antecedentes de hipotiroidismo congénito en tratamiento sustitutivo. Tiene una hermana de 3 años afecta de síndrome de Down. Acude a consulta de Atención Primaria por presentar desde hace 24 horas lesiones cutáneas consistentes en pápulas rojizas de pequeño tamaño, con vesícula central de contenido claro. Están distribuidas por tronco, ingles y zona del pañal. Tiene buen estado general, afebril, y no presenta rechazo de la ingesta, ni irritabilidad, ni otros signos asociados. La sospecha diagnóstica inicial es un exantema vírico inespecífico. Es remitida a su domicilio sin tratamiento y se indica observación de la evolución. Consulta de nuevo a las tres semanas por persistencia de las lesiones cutáneas, con la misma distribución corporal que la inicial, pero ya no son pápulo-vesiculares, sino que ahora presenta elementos pápulo-purpúricos (Figura 1). Presenta buen estado general, sigue afebril, no presenta ningún síntoma digestivo y en la exploración no se detecta ningún otro hallazgo de interés, ni adenomegalias, ni visceromegalias.
| Figura 1. Histiocitosis de células de Langerhans: lesiones pápulo-purpúricas en tronco anterior (izquierda) y posterior (derecha) |
|---|
 |

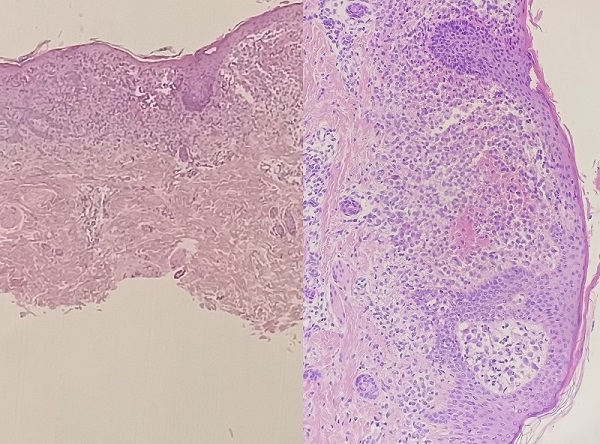
Ante la evolución inusual y la apariencia de las lesiones, se remite al Servicio de Dermatología, donde se decide realizar la biopsia de una de las lesiones. La anatomía patológica muestra proliferación de células que se extienden en sábana ocupando toda la dermis papilar, ulcerando la porción central de la epidermis. Son células ovoides de 15 micras de citoplasma eosinófilo y núcleo indentado concordantes con células de Langerhans, tanto histológica como inmunofenotípicamente, ya que expresan CD1a y S-100 de forma intensa. Se observan vasos dilatados y congestivos en dermis superficial, lo que daría la apariencia pápulo-purpúrica descrita macroscópicamente. El diagnóstico anatomopatológico es de histiocitosis de células de Langerhans (HCL) (Figura 2).
| Figura 2. Histiocitosis de células de Langerhans: anatomía patológica de una de las lesiones: imagen panorámica (izquierda) y en detalle celular (derecha) |
|---|
 |

Es derivada al Servicio de Hemato-oncología de referencia. En el estudio inicial para detectar otros órganos afectos, precisar la extensión y el grado de riesgo de la HCL, se descubre, además de la afectación cutánea, una infiltración del colon descendente. Se trata con el protocolo de quimioterapia de HCL multisistémica. Se consigue la remisión y así permanece desde hace 5 años.
DISCUSIÓN
La HCL constituye un grupo heterogéneo de enfermedades poco frecuentes, caracterizadas por la infiltración de casi cualquier órgano del cuerpo por células del sistema fagocítico mononuclear. Las hipótesis sobre su patogenia son dos y no son excluyentes: una excesiva respuesta inmune a un estímulo antigénico que causa una reacción inflamatoria desproporcionada o un proceso neoplásico. La HCL puede presentarse a cualquier edad, pero predomina por debajo de los 4 años. Tiene una incidencia anual de 4,6 casos por millón en menores de 15 años, con un predominio en varones de 1,2:11-4.
Su presentación clínica es muy variable. Puede afectar a solo un órgano de forma unifocal o multifocal (generalmente, lesiones óseas benignas o lesiones cutáneas inespecíficas) o ser multisistémica (2 o más órganos)1-5. La HCL es una de las enfermedades más simuladoras y puede confundirse inicialmente con muchas enfermedades; en el caso de las lesiones cutáneas, con infecciones víricas habituales3. La baja incidencia y el amplio espectro de manifestaciones clínicas a menudo provoca un retraso en el diagnóstico, que puede condicionar una extensión más amplia de la HCL, lo que provocaría la clasificación en un grupo de riesgo más alto y un peor pronóstico1,2.
La afectación ósea es la más frecuente y consiste en lesiones líticas en cráneo, fémur, cara, costillas o vértebras. Las lesiones cutáneas son las segundas más frecuentes. Otros órganos implicados pueden ser los ganglios linfáticos, la médula ósea, el bazo, el hígado, el pulmón, la glándula pituitaria y el sistema nervioso central. Los niños con afectación de médula ósea, hígado o bazo tienen peor pronóstico, con peor supervivencia a 5 años, y son clasificados como grupo de alto riesgo de HCL2,5,6.
La afectación gastrointestinal, como sucede en nuestro caso, solo se demuestra en el 2% de las HCL7. Puede afectar a cualquier parte del aparato gastrointestinal, desde la boca al ano, a veces es asintomática, como en nuestro caso, o con síntomas, siendo los más frecuentes la enteropatía pierde-proteínas, diarrea sanguinolenta, síndromes de malabsorción y fallo de medro7.
En nuestro caso, el diagnóstico se produjo al mes del inicio de las lesiones, pero cuando el debut es cutáneo el diagnóstico suele retrasarse una media de 6 meses, debido a la heterogeneidad de las lesiones, lo que dificulta el diagnóstico diferencial. La mediana de edad del comienzo de la lesiones es 3 meses (como en nuestro caso), y la del diagnóstico, 9 meses3,4.
Las lesiones cutáneas más frecuentes de la HCL son pápulas rosadas-amarillentas descamativas, con tendencia a la formación de costras en su superficie, localizadas característicamente en áreas seborreicas en el tronco, cara o cuero cabelludo8. Otra forma frecuente de presentación en niños muy pequeños es un exantema vesicular, que obliga a realizar el diagnóstico diferencial con miliaria, eccema, escabiosis o, incluso, varicela8. En recién nacidos y lactantes también puede presentarse como eccema seborreico o costra láctea persistente en cuero cabelludo, o bien con exantemas generalizados, con lesiones tipo pápulas, pústulas, vesículas o úlceras con costras hemorrágicas3. Otras formas de presentación cutáneas son lesiones similares a un exantema vírico inespecífico, dermatitis del pañal petequial, absceso frío posvacunal, pápulas marrones diseminadas, eccema seborreico purpúrico, granuloma piogénico, hemangioma y pápulas purpúricas3,4,9,10. La localización más frecuente es el tronco, seguido de la cabeza, ingles, pubis y axilas. La distribución en camiseta se considera patognomónica8. La afectación cutánea es única en el 12% de los casos de HCL. Cuando la HCL es multisistémica la piel se afecta en el 53% de los casos3,4,8-11.
El diagnóstico de la HCL se basa en la sospecha clínica, y se confirma mediante la biopsia de las lesiones accesibles. Se precisa de múltiples pruebas analíticas y radiológicas para establecer la extensión de la HCL, para conocer si es multisistémica y determinar el grado de gravedad, necesario para establecer el tratamiento más adecuado1-3,7.
El tratamiento de la HCL se adapta a la condición individual de riesgo. Los niños con lesiones únicas suelen responder al tratamiento local. En el caso de afectación única cutánea, se emplean inicialmente antisépticos y corticoides tópicos de potencia moderada o fuerte. Los pacientes con afectación cutánea extensa y sintomática o con enfermedad multisistémica requieren quimioterapia (inicialmente prednisona y vimblastina)2,3,5,11. En nuestro caso, es una HCL multisistémica y se trata con quimioterapia sistémica.
La supervivencia en pacientes sin disfunción orgánica es casi del 100%, pero la mortalidad cuando sí la hay puede alcanzar el 20%. A pesar de los avances en los tratamientos, la tasa de recidiva permanece por encima del 30%. El fallo de tratamiento inicial está asociado a una mayor mortalidad y a secuelas posteriores, como el temido síndrome neurodegenerativo (síndrome tardío, inicia entre 5 y 15 años después del diagnóstico, y progresivo, con afectación cerebelosa y troncoencefálica, que puede asociar deterioro neurocognitivo), secuelas ortopédicas (fracturas patológicas), cutáneas (ulceraciones y cicatrices) o endocrinológicas (diabetes insípida, hipotiroidismo o pubertad precoz)2,3,5,6,11. Nuestro caso permanece en remisión y sin secuelas a los 5 años del diagnóstico.
CONCLUSIÓN
Como conclusión, queremos incidir en la dificultad diagnóstica que supone la presentación inicial de la HCL con lesiones cutáneas porque pueden ser muy diversas. Se precisa un alto índice de sospecha para lograr un diagnóstico precoz que, junto con el enfoque multidisciplinario para el manejo adecuado de las formas multisistémicas de la HCL, proporcionan un mejor pronóstico tanto en la supervivencia como en la aparición de secuelas1-3,5.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
RESPONSABILIDAD DE LOS AUTORES
Todos los autores han contribuido de forma equivalente en la elaboración del manuscrito publicado.
Los autores han remitido un formulario de consentimiento de los padres/tutores para publicar información de su hijo/a.
ABREVIATURAS
HCL: histiocitosis de células de Langerhans.
BIBLIOGRAFÍA
- Emile JF, Cohen-Aubart F, Collin M, Fraitag S, Idbaih A, Abdel-Wahab O, et al. Histiocytosis. Lancet. 2021;398(10295):157-70. https://doi.org/10.1016/S0140-6736(21)00311-1
- Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med. 2018;379:856-68. https://doi.org/10,1056/NEJMra1607548
- Astigarraga I, García Obregón S, Pérez Martínez A, Gutiérrez Carrasco I, Santa María V, Rodríguez-Vigil C, et al. Histiocitosis de células de Langerhans. Avances en la patogenia y práctica clínica. Anales de Pediatría. 2022;97:130.e1-130.e7. https://doi.org/10.1016/j.anpedi.2022.05.002
- Morren MA, Broecke KV, Vangeebergen l, Sivellis-Smitt JH, van den Berghe P, Hauben E, et al. Diverse cutaneus presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-92. https://doi.org/10.1002/pbc.25834
- Rodríguez Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020;135:1319-31. https://doi.org/10.1182/blood.2019000934
- Allen CE, Ladisch SmcKlain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. https://doi.org/10.1182/blood-2014-12-569301
- Minkov M, Pötschger U, Thacker N, Astigarraga I, Braier J, Donadieu J, et al. Additive impact of gastrointestinal involvement in severe multisystem Langerhans cell histiocytosis. J Pediatr. 2021;237:65-70. https://doi.org/10.1016/j.jpeds.2021.06.016
- Valdivieso M, Bueno C. Histiocitosis de células de Langerhans. Actas Dermosifiliogr. 2025;96(5):275-84. https://doi.org/10.1016/S0001-7310(05)75054-7
- Afonso C, Dias T, Teixeira C, Rodrigues D, Mira M. Cutaneous manifestations on Langerhans cell histiocytosis in pediatric age: a case report. Cureus 16 (9): e70141. https://doi.org/10.7759/cureus.70141
- Khundadze M, Khurtsia l, Shulaia N, Kandelaki G. When histiocytosis masquerades as mononucleosis: a case report. Cureus. 16(9):e70156. https://doi.org/10.7759/cureus.70156
- Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-84. https://doi.org/10.1002/pbc.24367