
Vol. 23 - Num. 30
Casos clínicos. Miscelánea
La importancia de prestar atención al fenotipo clínico ya que a veces nos puede orientar al diagnóstico etiológico
Cristina Bardella Gila, Raquel Subirón Ortegob, Juan Hidalgo Sanzc, Inés Félez Molinerb, Rebeca Hernández Abadíac, César García Velad
aMIR-Pediatría. Hospital Universitario Miguel Servet. CS Torrero La Paz. Zaragoza. España.
bMIR-Pediatría. Hospital Universitario Miguel Servet. Zaragoza. España.
cServicio de Pediatría. Hospital Universitario Miguel Servet. Zaragoza. España.
dPediatra. CS Sagasta. Zaragoza. España.
Cómo citar este artículo: Bardella Gil C, Subirón Ortego R, Hidalgo Sanz J, Félez Moliner I, Hernández Abadía R, García Vela C. La importancia de prestar atención al fenotipo clínico ya que a veces nos puede orientar al diagnóstico etiológico. Rev Pediatr Aten Primaria. Supl. 2021(30):203-4.
Publicado en Internet: 30-11--0001 - Número de visitas: 6131
INTRODUCCIÓN
El síndrome de Marfan es una enfermedad hereditaria, sistémica del tejido conectivo que afecta a distintas estructuras (esqueleto, pulmones, ojos corazón, vasos sanguíneos). Por ello se caracterizada por una combinación variable de manifestaciones cardiovasculares, musculoesqueléticas, oftalmológicas y pulmonares.
Su prevalencia es 1/5000 y afecta igual en ambos sexos.
Los síntomas pueden aparecer a cualquier edad y son variables de una persona a otra, incluso dentro de una misma familia. La afectación más importante es a nivel cardiovascular ya que pueden presentar dilatación progresiva de la aorta, lo que afecta al pronóstico de la enfermedad.
CASO CLÍNICO
Niño inmigrante, de nacionalidad rumana, residente desde hace un año es España, en el que se objetiva en la revisión de los diez años una talla elevada muy superior al P99. Como único antecedente de interés, referían un hipospadias balano-prepucial que requirió cirugía a los dos años.
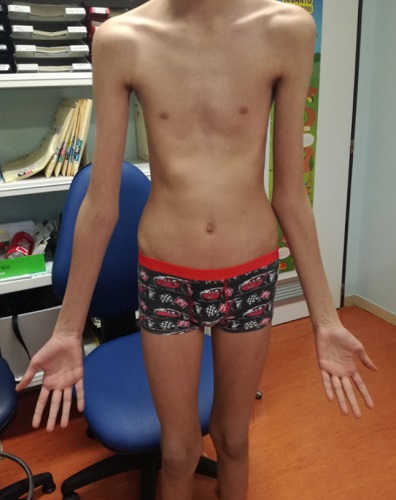
A la exploración se objetiva talla alta (altura de 183 cm, P>99 por edad y sexo), paladar ojival, pectus excavatum, aracnodactilia e hiperlaxitud articular (Fig. 1). Ante estos hallazgos, se deriva a la consulta de Endocrinología Pediátrica ante sospecha de patología del tejido conectivo.
| Figura 1. Imagen en la que se observa el fenotipo tan característico de nuestro paciente |
|---|
 |
En consultas, realizan estudio genético con cariotipo 46 XY y estudio genético dirigido a hiperlaxitud articular, identificando una variante patogénica en heterocigosis en gen FBN1 c.247+1G>A, con patrón de herencia autosómica dominante asociada al síndrome de Marfan.
CONCLUSIONES
La afectación más importante en los pacientes con un síndrome de Marfan es a nivel cardiovascular (ya que pueden presentar dilatación progresiva de aorta). Habitualmente, la afectación esquelética es el primer signo de la enfermedad (incluye talla alta, dolicostenomelia, escoliosis, aracnodactilia, escoliosis, deformidad torácica con pectus carinatum o excavatum, dolicocefalia, hiperlaxitud articular o hipoplasia malar). La afectación oftalmológica es la ectopia o luxación del cristalino y/o una miopía axial. También tienen un riesgo aumentado de neumotórax.
La mayoría de los casos, se debe a mutaciones en el gen FBN1 (15q21), que codifica para la fibrilina-1 (una proteína esencial del tejido conectivo). La herencia es autosómica dominante. El manejo debe de ser multidisciplinar.
La afectación aórtica condiciona el pronóstico por lo que el objetivo principal es limitar la dilatación aórtica (con betabloqueantes, limitación de deportes de riesgo) y seguimiento periódico. Con un seguimiento regular y un manejo adecuado, los pacientes presentan una esperanza de vida cercana a la de la población general.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.