

Quistes pancreáticos congénitos: revisión a propósito de un caso
María Portillo Sánchez-Portala, David Coca Robinotb, Miguel Rasero Ponferradac, Iván Carabaño Aguadod, Enrique Salcedo Lobatod, Marta Germán Díaze, Enrique Medina Benítezd
aSección de Gastroenterología, Hepatología y Nutrición infantil. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
bSección de Radiología Infantil. Servicio de Radiodiagnóstico. Hospital Universitario 12 de Octubre. Madrid. España.
cServicio de Radiodiagnóstico. Hospital Universitario 12 de Octubre. Madrid. España.
dSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
eSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Hospital Universitario 12 de Octubre. Madrid. España.
Correspondencia: M Portillo. Correo electrónico: maria.portillo@salud.madrid.org
Cómo citar este artículo: Portillo Sánchez-Portal M, Coca Robinot D, Rasero Ponferrada M, Carabaño Aguado I, Salcedo Lobato E, Germán Díaz M, et al. Quistes pancreáticos congénitos: revisión a propósito de un caso. Rev Pediatr Aten Primaria. 2019;21:e87-e89.
Publicado en Internet: 20-06-2019 - Número de visitas: 14490
Resumen
Los quistes pancreáticos congénitos son una entidad muy poco frecuente en Pediatría. Se desconoce su causa. Generalmente, son asintomáticos y suponen un hallazgo radiológico casual. Se suelen localizar en la cola o en el cuello del páncreas. Lo más frecuente es que sean lesiones aisladas. De forma puntual se han descrito asociadas a otras enfermedades, como el síndrome de De Jeune, el síndrome de Beckwith-Wiedeman o la enfermedad de von Hippel-Lindau. Su tratamiento clásico ha sido la cirugía. No obstante, dada la benignidad de estas lesiones en la infancia, así como la complejidad que supone el tratamiento quirúrgico, se aconseja un manejo conservador de los mismos.
Palabras clave
● Múltiple ● Páncreas ● QuistesINTRODUCCIÓN
Los quistes pancreáticos congénitos son una entidad muy poco frecuente en Pediatría, por lo que implican un desafío diagnóstico1. Su etiología es desconocida, aunque se presupone que son el resultado de una alteración en la embriogénesis de los conductos pancreáticos2. Generalmente, son asintomáticos y suponen un hallazgo casual en una prueba de imagen realizada por otro motivo1. Su tratamiento clásicamente ha sido quirúrgico, a través de una exéresis completa de la lesión2. Recientemente se está preconizando un manejo conservador. En este sentido, desde el marco de la Atención Primaria, la divulgación del siguiente caso puede ser de interés, pues una vez establecido el diagnóstico, podría optarse por un seguimiento ambulatorio periódico, realizado desde la proximidad al paciente.
CASO CLÍNICO
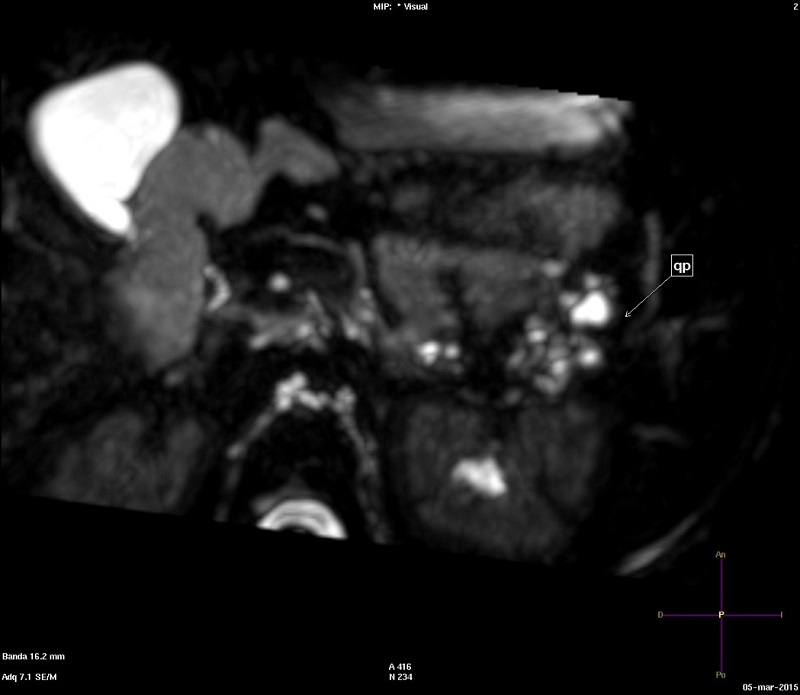
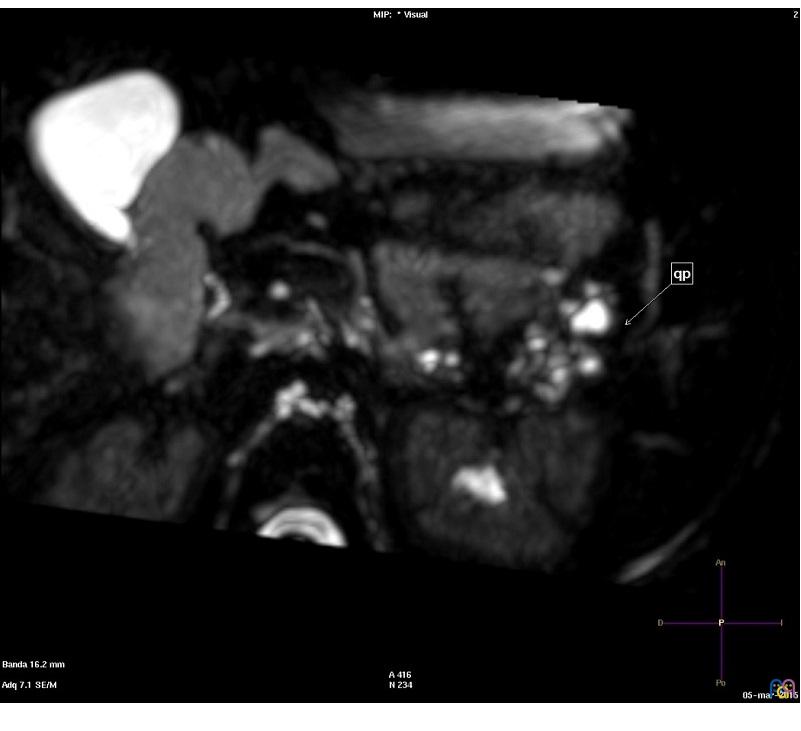
Niña de cinco años con un síndrome polimalfortativo consistente en médula anclada intervenida, quistes renales, polidactilia y retraso psicomotor, con diagnóstico genético de cromosomopatía por duplicación 13q31.1-q33.2 y deleción 13qter. La niña fue derivada a consultas externas de Digestivo por el hallazgo casual de quistes pancreáticos. Los quistes pancreáticos fueron objetivados por primera vez en ecografía abdominal a los dos años de vida como parte del estudio del cuadro polimalformativo, hallándose un páncreas ligeramente aumentado de tamaño, así como múltiples quistes a nivel de la cola y el cuerpo de alrededor de 10 mm. La paciente se encontraba asintomática desde el punto de vista gastrointestinal. Normalidad analítica, incluyendo amilasa y lipasa. Se realizaron controles ecográficos seriados, así como una resonancia magnética a los cinco años, en la que se observó disminución del tamaño de los quistes, siendo el mayor de ellos de 3 mm y limitándose a la cola pancreática (Fig. 1). Se decidió mantener actitud expectante, dado su carácter asintomático y el tamaño pequeño de los mismos.
| Figura 1. Resonancia magnética en plano axial con secuencia colangiográfica potenciada en T2, en la que se reconocen múltiples quistes hiperintensos en la cola pancreática (marcado con una flecha, y rotulado como "qp") |
|---|
 |
DISCUSIÓN
Los quistes pancreáticos constituyen una entidad extremadamente rara en niños, aunque relativamente frecuente en adultos. Se clasifican en seis grupos1: 1) congénitos; 2) retentivos; 3) de duplicación; 4) pseudoquistes; 5) neoplásicos, y 6) parasitarios. Los quistes congénitos, retentivos y de duplicación son considerados como quistes verdaderos y su diferencia frente a los pseudoquistes radica en que los primeros están delimitados por un epitelio que es capaz de secretar fluidos, mientras que los pseudoquistes se constituyen por una capa fina de tejido fibrótico-inflamatorio que se forman secundariamente a traumatismos, inflamación o infecciones2. Es difícil estimar su incidencia, pero los pseudoquistes se consideran mucho más frecuentes que los quistes verdaderos y, dentro de los mismos, los quistes congénitos son los más infrecuentes de todos, suponiendo menos de un 1% del total.
Se desconoce la etiología de los quistes congénitos, aunque se presupone que son la consecuencia de una alteración en el desarrollo del sistema ductal pancreático. Durante el desarrollo embriológico, se forman unos conductos que posteriormente son reemplazados por los definitivos. La alteración en la regresión de estos conductos puede dar lugar a la formación de quistes pancreáticos congénitos. Sin embargo, los quistes de retención se forman postnatalmente como consecuencia de la obstrucción de una parte del sistema ductal2. Histológicamente, es difícil diferenciarlos y puede ser útil el análisis bioquímico del líquido intraluminal, ya que en el caso de los quistes congénitos el nivel de amilasa suele ser bajo3.
Los quistes pancreáticos pueden encontrarse en cualquier punto del órgano, pero la localización más frecuente de los mismos es la cola y el cuello (62%)3.
Aunque en la mayoría de las ocasiones se presentan de forma aislada, pueden aparecer hasta en el 30% de los casos asociados a otras entidades como pueden ser el síndrome de De Jeune o displasia torácica asfixiante, polidactilia, síndrome de Beckwith-Wiedeman, enfermedad de von Hippel-Lindau, malformaciones anorrectales, síndrome de Ivermark, ectasia tubular renal y poliquistosis renal3. Los quistes pancreáticos suelen ser únicos, pero cuando aparecen en el contexto de algunas entidades previamente nombradas como la enfermedad de Von Hippel-Lindau o la poliquistosis renal, pueden ser múltiples.
Los quistes pancreáticos suelen ser asintomáticos y en la mayoría de los casos son diagnosticados como hallazgo casual en prueba de imagen abdominal realizada por otro motivo. En ocasiones, sobre todo si son de gran tamaño, pueden producir clínica secundaria a la compresión de órganos adyacentes. Las complicaciones, como sobreinfección o inflamación, son extremadamente raras, aunque posibles4.
El diagnóstico prenatal de estos quistes no es fácil, pues la visualización completa del páncreas es técnicamente difícil en el seguimiento ecográfico gestacional5. Suelen diagnosticarse en los primeros dos años de vida, como ocurrió en el caso previo descrito. Los análisis de laboratorio suelen ser normales, salvo complicaciones como sobreinfección o inflamación.
A pesar de los avances experimentados en los últimos años con las pruebas de imagen, el diagnóstico continúa siendo complejo. La ecografía abdominal es útil para diferenciar lesiones quísticas con contenido líquido intralesional, de lesiones sólidas pancreáticas. Sin embargo, en muchas ocasiones no permite evaluar el verdadero origen de la lesión y se precisan otras técnicas como la tomografía computarizada o la resonancia magnética, siendo esta última superior. La colangiopancreatografía retrógrada endoscópica es una técnica invasiva que permite objetivar si existe comunicación con el conducto biliar o pancreático, pero no se realiza de rutina2.
Clásicamente, el tratamiento de los quistes pancreáticos congénitos ha sido la escisión completa. Hoy se reserva solo para casos sintomáticos o complicados. El tipo de cirugía varía en función del número de quistes y su localización. Los quistes localizados en cuerpo y cola pueden ser extirpados mediante pancreatectomía distal, mientras que en los que se localizan en la cabeza del páncreas existen más dificultades técnicas, precisando cistoyeyunostomía en Y de Roux o incluso son susceptibles de drenaje por vía interna6. Una vez extirpados o drenados, los quistes no suelen recidivar en el seguimiento posterior de los pacientes. Estas técnicas no están exentas de riesgos y aunque la mortalidad está por debajo del 1%, la incidencia de complicaciones no es despreciable7. Por ello, dada la benignidad de las lesiones quísticas en edad pediátrica y la complejidad que supone el tratamiento quirúrgico, cada vez es más frecuente un manejo conservador de los mismos con actitud expectante. La opción combinada de seguimiento entre Atención Primaria y hospital puede ser una opción razonable.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
BIBLIOGRAFÍA
- Basturk O, Cohan I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis, and clinical implications. Arch Pathol Lab Med. 2009;133:23-38.
- Shahid M, Javed Z, Usman M, Iltaf S. True congenital pancreatic cyst: a rare entity. Cureus. 2018;10:3318.
- Al-Salem AH, Matta H. Congenital pancreatic cyst: diagnosis and management. J Pediatr Gastroenterol Nutr. 2014;59:38-40.
- Castellani C, Zeder SL,Spuller E, Hollwarth ME. Neonatal congenital pancreatic cyst: diagnosis and management. J Pediatr Surg. 2009;44:1-4.
- Chung JH, Lim GY, Song YT. Congenital true pancreatic cyst detected prenatally in neonate: a case report. J Pediatr Surg. 2007;42:27.
- Gentimi FE, Papandreou E, Tzovaras AA. Pancreatic cystic lesion in an infant. J Indian Assoc Pediatr Surg. 2011;16:72-4.
- Bawazir OA, Al-Salem AH, Bawazir AO. Congenital pancreatic cyst: preoperative diagnosis and management. J Pediatr Surg Case Reports. 2017;21:16-21.