
Vol. 11 - Num. 16
Endocrinología pediátrica
Síndrome metabólico
Sara Artola Menéndeza, Mar Duelo Marcosb, Eva Escribano Cerueloc
aMédico internista. CS Hereza, Servicio Madrileño de Salud, Área 9. Madrid. España.
bPediatra. CS Las Calesas. Madrid. España.
cPediatra. Hospital Universitario Niño Jesús. Madrid. España.
Cómo citar este artículo: Artola Menéndez A, Duelo Marcos M, Escribano Ceruelo E. Síndrome metabólico. Rev Pediatr Aten Primaria. 2009;11 Supl 16:s259-s277.
Resumen
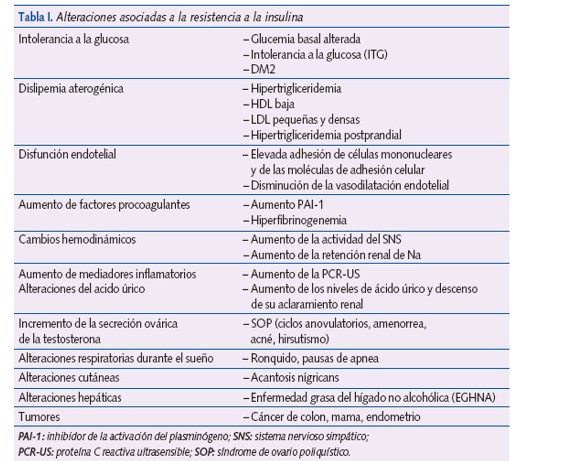
La obesidad infantil es el principal factor de riesgo de obesidad en el adulto, así como de síndrome metabólico, diabetes mellitus tipo 2 y del desarrollo de enfermedades cardiovasculares, que reducen la calidad y duración de la vida. El síndrome metabólico es un diagnóstico clínico que identifica una población con elevado riesgo cardiovascular.
La resistencia a la insulina parece ser el vínculo común y el factor promotor de la cascada de disturbios metabólicos, modulados por factores genéticos y ambientales.
El síndrome metabólico puede demostrarse en la edad pediátrica, pero actualmente no existen criterios claramente definidos. Los criterios de la International Diabetes Federation (IDF) para niños y adolescentes son los más utilizados, por su fácil manejo; la medida de la cintura es el principal componente. Se necesita una definición internacional para poder comparar los resultados de los diferentes estudios, y, hasta que no ocurra, no se debe diagnosticar por debajo de los 10 años.
Palabras clave
● Adolescentes ● Diabetes mellitus tipo 2 ● Niño ● Obesidad ● Resistencia a la insulina ● Síndrome metabólicoIntroducción
La obesidad infantil es el principal factor de riesgo para el desarrollo de obesidad en el adulto, así como de síndrome metabólico (SM), diabetes mellitus tipo 2 (DM 2) y de enfermedades cardiovasculares (ECV) que disminuyen la calidad y duración de vida.
El SM fue descrito por Reaven en 1988 como síndrome X. Otras denominaciones utilizadas, no todas superponibles, son: síndrome plurimetabólico, cuarteto de la muerte, sexteto de la muerte, etc.
El SM es un diagnóstico clínico que identifica una población con elevado riesgo CV (RCV). Sin embargo, esta población es heterogénea, ya que las diferentes asociaciones de los elementos del SM tienen distinto valor predictivo de ECV; así, un adolescente con perímetro de cintura (PC) > p90, triglicéridos (TG) elevados, y colesterol HDL bajo, no tiene el mismo RCV que otro con hiperglucemia > 100 mg/dl, presión arterial (PA) > p90 y PC > p90, aunque ambas situaciones se consideren SM.
Parece que la resistencia a insulina (RI) es el vínculo común y el factor promotor de la cascada de disturbios metabólicos, modulados por factores genéticos y ambientales1. Las anomalías metabólicas presentes en la obesidad se asocian más con la distribución de la grasa que con el grado de obesidad. La grasa intraabdominal altera los mecanismos inflamatorios y promueve la RI. La disminución de adiponectina (fundamentalmente) y el aumento de leptina pueden ser los mediadores iniciales de la RI2.
Fisiopatología del síndrome metabólico y de la resistencia a la insulina
Durante mucho tiempo el tejido adiposo ha sido considerado un reservorio de energía en forma de triglicéridos (TG). En la última década se ha reconocido su importancia como órgano endocrino secretor de adipokinas (AK), con actividad local y sistémica (SNC, músculo esquelético, hígado y huesos) que modulan la ingesta y regulan el metabolismo energético, sobre todo el lipídico, pero también el hidrocarbonado3,4 (figura 1). Además, participan en los procesos de inflamación, enfermedad vascular ateroesclerosa y cáncer5.
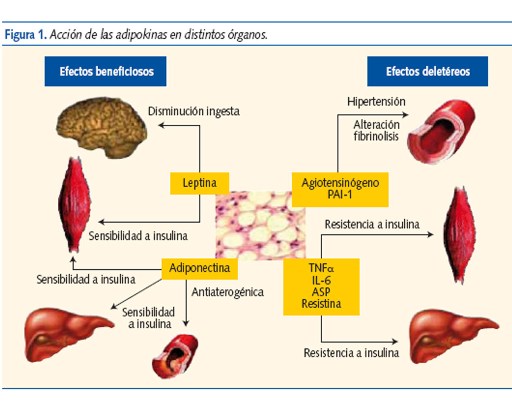
La regulación de la síntesis de AK por el adipocito es compleja y en gran parte desconocida. Su liberación está regulada por diferentes hormonas, como la insulina y la hormona de crecimiento, y algunos autores proponen la existencia de un eje hipotálamo-hipófiso-adipocitario6.
En la obesidad visceral está aumentada la lipolisis, produciéndose un acúmulo de ácidos grasos (AG) libres y un cambio de la secreción de AK con alteraciones metabólicas que favorecen el desarrollo y mantenimiento de la obesidad y de sus complicaciones: RI, DM 2, SM y ECV.
La RI se define como la disminución de la capacidad de la insulina para ejercer sus acciones en tejidos diana: músculo esquelético, hígado o tejido adiposo. La RI, el SM y la aterosclerosis parecen tener una base inflamatoria común; se admite que la RI es el proceso fisiopatológico que subyace bajo el conjunto de factores de RCV del SM.
La insulina activa su receptor en la membrana celular (figura 2) y genera “segundos mensajeros” (insulin receptor substrate (IRS), Shc, GAB-1…); que estimulan dos vías de acción.
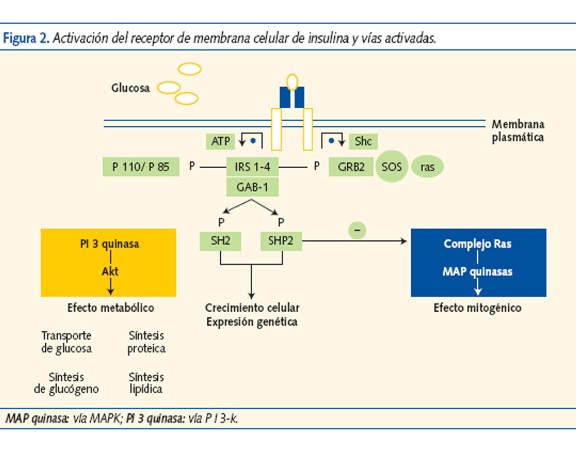
La vía PI-K (cascada de reacciones de fosforilación-3-quinasa), favorece el transporte de glucosa al interior de la célula y la vía MAPK, que promueve el crecimiento, la diferenciación y la proliferación celular. La RI altera la vía PI-K y disminuye el transporte y la utilización de glucosa. La hiperinsulinemia compensadora sobreestimula la vía MAPK que potencia la acción mitogénica y la migración de las células musculares lisas de los vasos, favoreciendo la aterogénesis (figura 3).
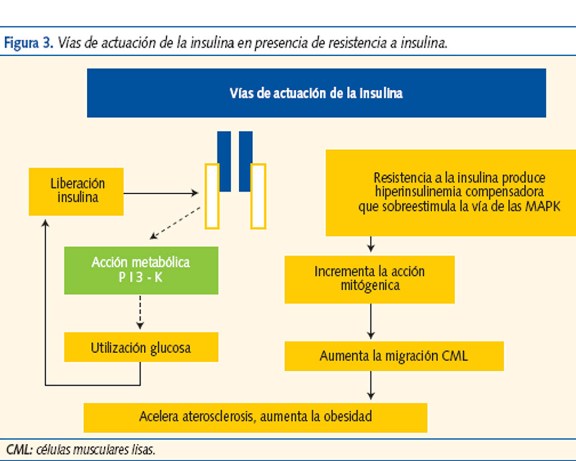
La relación entre RI y aterogénesis se produce por diferentes mecanismos: dislipemia, elevación del inhibidor del activador del plasminógeno (PAI-1), del fibrinógeno y del tono vascular, disfunción endotelial, etc.
La hiperinsulinemia inicial sería suficiente para mantener los niveles de glucemia pero, si se acompaña en su evolución de insuficiente secreción de insulina, aparecería intolerancia a la glucosa (ITG) y finalmente DM 2.
En los niños obesos existe un cambio en la secreción de AK: disminuye la adiponectina y la leptina aumenta, ambas alteraciones (sobre todo la primera) pueden ser los mediadores iniciales de la RI2.
La leptina se sintetiza fundamentalmente en el tejido adiposo subcutáneo, su nivel se correlaciona con los depósitos grasos, ya que aumenta con el índice de masa corporal (IMC) y la masa grasa, y disminuye con el ayuno. Se considera la principal hormona liporreguladora para una correcta homeostasis lipídica intracelular7. Su principal función es informar al SNC de la cantidad de grasa corporal, de manera que la ingesta, el metabolismo y la fisiología endocrina se adapten al estado nutricional. Por acción directa sobre el hipotálamo, produce estímulos anorexígenos: estimula la pérdida de peso al disminuir el apetito e incrementar el metabolismo8. En la obesidad exógena no parece estar alterado el gen de la leptina, aunque suele existir hiperleptinemia por insensibilidad. Solo en algunos casos de obesidad mórbida se ha descrito una alteración genética por déficit de leptina9.
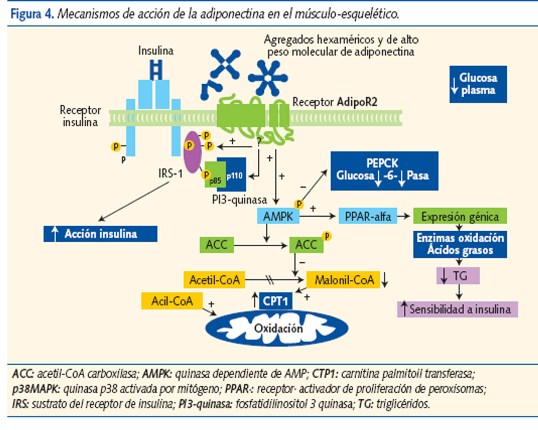
La adiponectina se secreta exclusivamente en el tejido adiposo. Mejora el control de la glucemia y tiene funciones hipolipemiantes, antiinflamatorias y antiaterogénicas. Actúa sobre receptores hepáticos (adipoR1) y del músculo esquelético (adipoR2); aumenta la sensibilidad a la insulina, estimula la oxidación de los AG, suprime la neoglucogénesis hepática e inhibe la adhesión de monocitos10. Activa las rutas catabólicas e inhibe las anabólicas11, actuando como un sensor de energía en la célula. Modula la respuesta inflamatoria de las células endoteliales y evita la transformación de macrófagos en células espumosas (crucial para el desarrollo de ateromatosis).
La adiponectina aumenta la sensibilidad a la insulina, activando los receptores PPAR-a (activador de proliferación de peroxisomas) que disminuyen los TG, modulan el estímulo de insulina y activan la AMP quinasa (figura 4). El efecto sobre el músculo esquelético es un menor contenido de TG; uno de los factores que aumentan la sensibilidad a la insulina, y que sugiere que la adiponectina protege de la RI y de la obesidad.
La hipoadiponectinemia se correlaciona más con la hiperinsulinemia y la RI que con el grado de adiposidad. La disminución de adiponectina depende de la distribución grasa corporal, y es el nexo de unión entre adiposidad, RI y metabolismo lipídico. Se ha demostrado que la hipoadiponectinemia predice el riesgo de padecer DM2, incluso en ausencia de otros indicadores de RI, y podría ser un marcador clave en el diagnóstico de SM, incluso en niños12.
La adiponectina está disminuida en gran variedad de estados patológicos asociados a RI: obesidad, DM2, HTA, ECV y SM.
Se cree que la hipoadiponectinemia ocurre por la inhibición del factor de transcripción PPAR-gamma13. Las tiazolidindionas (agonistas del PPAR-gamma) usadas en el tratamiento de la DM2, mejoran la tolerancia a la glucosa y la RI, aumentando la secreción de adiponectina14, aunque en la actualidad no se utilizan debido a sus efectos secundarios.
En la obesidad existe un estado de inflamación de bajo grado, caracterizado por niveles altos de PCR ultrasensible y de AK proinflamatorias: IL 1,6 y 18 o TNF-a, el fibrinógeno, el factor inhibidor del plasminógeno y por niveles bajos de AK antiinflamatorias, como la adiponectina.
La lista de AK ha ido creciendo, y existen indicios de que será aún mayor: adipsina, resistina, MCP-1, visfatina, etc. Su conocimiento ayudará a entender la complejidad del tejido adiposo como órgano endocrino.
Las AK podrían ser marcadores aún más precoces que los parámetros bioquímicos clásicos del SM, pero falta por determinar si estas alteraciones son consecuencia de la propia obesidad o si existe una predisposición genética que modifique su expresión y favorezca el desarrollo del SM desde la infancia.
Definición del SM
El SM es un diagnóstico clínico; un grupo de datos clínicos que aumentan el riesgo de ECV y de DM 2. La influencia en el RCV de cada factor es diferente en cada individuo. La RI y la obesidad abdominal son los ejes centrales del síndrome que aparece en individuos metabólicamente susceptibles15.
Desde la descripción inicial de Reaven se han propuesto diferentes definiciones para adultos (tabla II). Actualmente, las más aceptadas son las de la National Colesterol Education Program (NCEP)16 y la de la International Diabetes Federation (IDF)17.
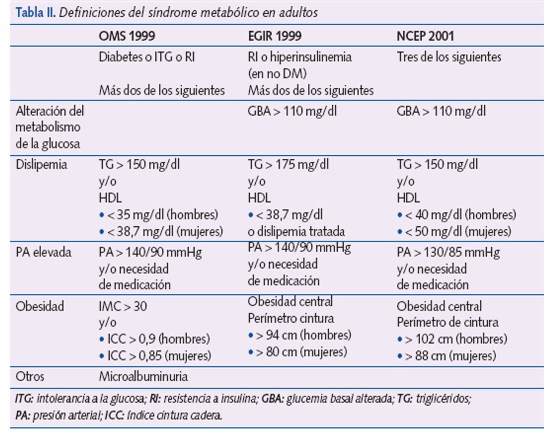
El SM puede demostrarse en la edad pediátrica, pero actualmente no existen criterios claramente definidos. En la tabla III se encuentran las diferentes definiciones del SM en la edad pediátrica. Cook et al.18 modificaron los criterios utilizados en los adultos para su uso en adolescentes.
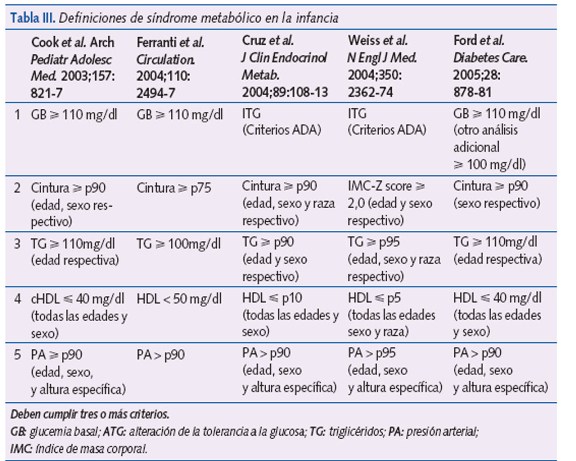
Pero en los últimos años se utilizan preferentemente los criterios de la IDF19 para niños y adolescentes (tabla IV); por su fácil manejo y, además, porque la medida de la cintura es el principal componente (potente predictor de RI). Divide a los niños en grupos de edad. Entre los 10-15 años hay criterios diagnósticos específicos, y por encima de los 16 años se utilizan los criterios para adultos.

Se necesita una definición internacional, para poder comparar los resultados de los diferentes estudios, y hasta que no ocurra no se puede hablar de esta entidad por debajo de los 10 años.
Aspectos epidemiológicos del SM
La prevalencia en países occidentales varía del 15 al 25% de la población general, aunque existe gran variabilidad al no haberse aceptado una definición universal.
El III National Health and Nutrition Examination Survey20 (NAHNES) 1988-1994, estudió la prevalencia del SM en 2.340 adolescentes americanos entre 12 y 19 años. Se diagnosticó SM en: el 4,2% (6,1% de los varones y 2,1% de las mujeres); el 28,7% en los obesos; el 6,8% de los chicos con sobrepeso y el 0,1% de los chicos con un IMC < p85. Estas prevalencias han aumentado de forma paralela a la obesidad y han alcanzado el 50% en adolescentes con obesidad grave21.
Con los nuevos criterios de la IDF para adolescentes, la prevalencia de SM en el estudio NAHNES es del 9,4% en los niños y del 9,7% en las niñas.
En Europa, la prevalencia de SM en niños es variable, desde un 33% en el Reino Unido y un 27% en Turquía, hasta un 9% en Hungría22.
En España se ha encontrado una prevalencia de SM del 18% (utilizando los criterios de Cook et al.18) y de RI del 35% en niños con obesidad moderada entre 4 y 18 años23.
Relación entre SM y ECV
El SM triplica el riesgo de cardiopatía coronaria o enfermedad cerebrovascular.
El impacto que el SM tiene en la población adulta sobre la ECV y muerte no se manifiesta en la edad pediátrica, aunque el proceso patológico y los factores de riesgo se inicien en ella. Como las alteraciones metabólicas tienden a persistir en el tiempo (tracking), hay una alta probabilidad de que los niños obesos se conviertan en adultos obesos con dislipemia, por lo que tienen especial riesgo de aterogénesis y de ECV.
Se estima que el 77% de los niños obesos serán adultos obesos y, es probable, que la persistencia de los factores de riesgo de ECV a lo largo de la infancia sea la que confiera dicho riesgo, más que la misma obesidad. Estudios realizados en cohortes amplias de individuos24 observan que a mayor IMC en la infancia mayor riesgo de enfermedad coronaria en el adulto; riesgo que aumenta con la edad del niño y que es mayor en niños que en niñas. Aunque no se ha podido establecer puntos de corte del IMC a partir de los cuales haya un aumento brusco del riesgo.
Diabetes mellitus tipo 2 (DM2)
Hasta hace pocos años era excepcional en la etapa infantil y adolescente. Pero con el incremento de la obesidad, la prevalencia de la DM2 ha aumentado, sobre todo en la pubertad. El riesgo de desarrollar DM2 aumenta con la duración e intensidad de la obesidad.
Aunque todos los obesos tengan algún grado de RI, no todos desarrollan ITG. Actualmente no se conocen totalmente los factores que influyen. Existe una fuerte predisposición familiar y en un futuro próximo, los marcadores genéticos podrán ayudar a identificar a los hijos de padres diabéticos con mayor riesgo de desarrollar diabetes. Goran et al.25 estudiaron 150 niños latinos con sobrepeso e historia familiar de DM2; se encontraron ITG en el 28% (independientemente del grado de obesidad); y en el 41% de los niños con antecedente de DM gestacional.
En niños y adolescentes la DM2 suele ser asintomática, aunque puede haber algunos hallazgos casuales (glucosuria) o inespecíficos (candidiasis vaginal). La prevalencia de la DM2 en la población infantil no está bien establecida, se estima que una importante proporción de los niños obesos (21-28%) se encuentran en situación de prediabetes26. La prediabetes se caracteriza por una glucemia en ayunas elevada (110-125 mg/dl) y/o ITG; en adultos se considera un estadio intermedio en la evolución a DM2, pero aún no está claro que los niños que la presenten tengan el mismo riesgo de progresión. De sus dos componentes en los niños obesos, es más habitual la ITG que el aumento de la glucemia en ayunas. Sinha et al.21 encontraron que el 25% de 55 niños obesos y el 21% de 112 adolescentes obesos tenían ITG con valores de glucemia a las 2 horas de la sobrecarga oral de glucosa > 140 mg/dl y el 4% tenían DM2 no diagnosticada.
Como la alteración de la glucemia puede permanecer asintomática años, la ADA ha publicado unas recomendaciones para su despistaje (tabla V)27.

Alteraciones lipídicas en la obesidad
Forman parte de los criterios del SM y de los factores de RCV.
La dislipemia en los niños obesos se caracteriza por lo siguiente: aumento de TG, del colesterol total, del LDL y de la apolipoproteína B, con disminución de HDL y de la ApoA-1. Pero la tríada más importante incluida en las definiciones de SM en la infancia incluye: aumento de TG, de partículas LDL (especialmente las densas y pequeñas) y descenso de HDL. Varios estudios longitudinales28-30 encontraron que estos tres parámetros eran los más fiables para predecir el RCV en el adulto joven, además de la HTA y la obesidad.
Estudios recientes han evaluado el grosor de la carótida por ultrasonidos en 879 niños de 3 a 18 años, hasta los 24-39 años y encontraron que el perfil de apolipoproteínas B y A1 (ApoB/ ApoA1 elevado) predice mejor los daños aterogénicos que el nivel de HDL y LDL31. Pero Frontini et al.32 al analizar los datos del estudio Bogalusa, concluyen que la capacidad de previsión del RCV de las apolipoproteínas es inferior a la determinación del colesterol no HDL, que incluye las LDL, VLDL y las lipoproteínas de densidad intermedia; de confirmarse, esto abarataría el cribado de dislipemia.
HTA en la obesidad
Numerosos estudios han confirmado la asociación entre obesidad y HTA, lo que forma parte de los criterios del SM y de los factores de RCV.
El hiperinsulinismo favorece la HTA por varios mecanismos: depósito de lípidos en las arterias, hipertrofia de la íntima del endotelio, vasoconstricción por estimulación del sistema simpático y retención renal de sodio.
Diferentes estudios epidemiológicos han mostrado que el riesgo de mantener los percentiles de PA desde la infancia a la edad adulta es alto. Pero existen múltiples mecanismos ambientales y genéticos que influyen en el desarrollo de la HTA, por lo que es difícil valorar en qué medida la obesidad y/o la hiperinsulinemia influyen en su génesis.
La HTA es reversible con la pérdida de peso.
Enfermedad grasa del hígado no alcohólico (EGHNA)
Se sospecha cuando se detectan alteraciones bioquímicas hepáticas y cambios en las pruebas de imagen sugerentes de depósito anormal de grasa. La obesidad es su principal causa y se está convirtiendo en una de las hepatopatías más frecuentes33.
Al ser un diagnóstico histológico es difícil conocer su prevalencia real. En un estudio americano de fallecidos accidentalmente entre 2 y 19 años se diagnosticó en el 9,6%. Según el peso, presentaron afectación hepática el 5% de los niños con peso normal, el 16% de los que tenían sobrepeso y el 38% de los niños obesos34.
Se sospecha por las cifras elevadas de aminotransferasas y por ecografía hepática (que detecta esteatosis cuando al menos están afectados el 30% de los hepatocitos). La mayoría están asintomáticos, y se detecta al evaluar el sobrepeso-obesidad. Es más frecuente en varones entre 11-13 años y altos para su edad. Algunos niños refieren dolor en hipocondrio derecho. Puede haber hepatomegalia, pero el dato es difícil de constatar por el abundante panículo adiposo. Solo un 10% de los niños tienen peso normal.
Las aminotransferasas se elevan de forma ligera o moderada, con cifras entre 2 a 10 veces el valor normal. Puede existir aumento de la gammaglutamil transpeptidasa.
La biopsia es imprescindible para evaluar la repercusión hepática y confirmar el diagnóstico. Existe un score para graduar las lesiones de EGHNA, desde esteatosis simple a EGHNA clasificada de 1 a 4, según el grado de inflamación y fibrosis y, en último término, cirrosis y hepatocarcinoma.
No se conoce la etiología, pero se asocia a obesidad, DM2 e hiperlipemia. Prácticamente todos los niños tienen RI, que parece ser el mecanismo que conduce a la EGHNA. Su interés radica en la posibilidad de evolucionar hacia formas de hepatopatía severa. Es una patología relativamente nueva: no se conocen los determinantes para el paso de simple esteatosis sin trascendencia vital, a la forma potencialmente progresiva.
No hay tratamiento eficaz. Se recomienda controlar la obesidad, la DM2 y la hiperlipemia.
Recomendaciones terapéuticas
El objetivo del tratamiento no es el SM en sí mismo, sino el de cada uno de sus componentes (factores de RCV). El SM no predice un mayor riesgo de futuras complicaciones que el que predice individualmente cada uno de sus componentes.
Es importante implicar a la familia y proporcionar información sobre la obesidad, sus complicaciones y cómo debe ser el tratamiento. Se utilizan tratamientos psicológicos cognitivo-conductuales, para ayudar a modificar los hábitos alimentarios y de actividad física.
La alimentación tiene como objetivo mejorar la sensibilidad a la insulina y prevenir o corregir las alteraciones metabólicas y CV. Debe ser equilibrada con un 50% de hidratos de carbono, un 30% de grasas y un 20% de proteínas. Distribuir las comidas en tres comidas principales y dos secundarias (media mañana y merienda).
Los alimentos con índice glucémico bajo mejoran la sensibilidad a la insulina y las alteraciones de los lípidos proporcionan más fibra, mayor sensación de saciedad y disminuyen la ingesta35. Las proteínas deben ser de alto valor biológico, y las grasas incluir AG esenciales y vitaminas liposolubles.
La actividad física tiene como objetivo mejorar la sensibilidad a la insulina, disminuir la lipogénesis, aumentar el gasto de energía y la pérdida de peso. Hay que cambiar los hábitos del niño y que la actividad física sea parte de su vida diaria: paseos (ir al colegio andando o subir escaleras), participar en deportes escolares y actividades deportivas el fin de semana. Debe individualizarse y buscar la actividad que más le guste y que le motive.
Tratamiento con fármacos
Está indicado en la obesidad grave, pero aún limitado a la adolescencia.
El olistat (inhibidor de la lipasa pancreática), permitido a partir los 12 años, disminuye la absorción de grasas. Como efectos secundarios presenta: flatulencia, diarrea y déficit de vitaminas liposolubles. Es más eficaz si se asocia a dieta y aumento de la actividad física.
La sibutramina (inhibidor de la recaptación de serotonina y noradrenalina) disminuye el apetito y aumenta la saciedad. Los efectos secundarios que produce son: sequedad de boca, insomnio, palpitaciones, aumento de la PA, taquicardia y ansiedad. Aprobado a partir de los 16 años.
La dieta, el ejercicio físico regular y la disminución de peso mejoran la sensibilidad a la insulina y la glucemia. Pero si no se consigue controlar la glucemia, hay que iniciar tratamiento farmacológico. Si presenta cetosis, empezar con insulina y cuando se controle glucemia, sustituir por metformina (antidiabético oral biguanida), aprobado a partir de los 10 años. Aumenta la sensibilidad a la insulina, la utilización periférica de glucosa y disminuye la glucosa en el hígado. La EGHNA asociada a obesidad y las niñas con síndrome de ovario poliquístico (hirsutismo, obesidad e hiperandrogenismo) presentan buena respuesta a este fármaco. El efecto secundario más grave, aunque excepcional, es la acidosis láctica. La diarrea, que puede aparecer hasta en un 20% de los casos, es dependiente de la dosis; siempre se recomienda la titulación de la dosis.
En los pacientes que no presentan síntomas de hiperglucemia y que los cambios logrados con el ejercicio y la alimentación no consigan controlar la glucemia, hay que iniciar tratamiento con metformina y si con ella no se alcanza el control, con insulina glargina en una dosis nocturna. Si a pesar de ello continúa con hiperglucemia, añadir análogos de la insulina de acción rápida en las comidas principales.
Cuando el valor del colesterol unido a las lipoproteínas de baja densidad (LDL) sea superior a 190 ó 160 mg/dl y haya antecedentes familiares de ECV, o presente 2 o más factores de riesgo asociados, el tratamiento indicado son las resinas de intercambio iónico: el ezetimiba (inhibe la absorción de colesterol) que puede utilizarse en mayores de 10 años. Los inhibidores de la HMG-CoA aún no se recomiendan en la infancia, ya que no se han realizado estudios.
SM y retraso de crecimiento intrauterino (RCIU)
La fase más dinámica de crecimiento humano ocurre prenatalmente y cualquier insulto en este período afecta al crecimiento longitudinal, lo que puede tener repercusiones permanentes, que generalmente solo ocurren en niños con crecimiento rápido postnatal.
Los individuos con RCIU tienen RI y si postnatalmente hay un aumento rápido de peso y talla, se produce un déficit de captación de glucosa muscular, hiperinsulinismo y un aumento compensador de adipocitos (acumulo de grasa central). A los 8 años el grado de RI y la proporción de grasa abdominal en niños con sobrepeso y antecedente de RCIU es el doble que en niños con peso adecuado para la edad gestacional y con el mismo IMC36.
Las niñas con antecedente RCIU y recuperación postnatal rápida también presentan una adrenarquia fisiológica más exagerada y una prevalencia alta de disfunción ovulatoria e hiperandrogenismo ovárico subclínico (síndrome de ovario poliquístico), alteraciones asociadas a aumento de la grasa abdominal (sin obesidad), hiperinsulinismo y aumento de sulfato de dehidroepiandrosterona (marcador de la secreción de andrógenos suprarrenales); alteraciones que son reversibles con los sensibilizantes de la insulina, como la metformina37.
La pubarquia precoz (PP) es la causa más frecuente de adrenarquia precoz. La PP constituye un factor de riesgo para el desarrollo de hiperandrogenismo ovárico en la adolescencia, fundamentalmente las pacientes con bajo peso al nacer y aumento rápido del IMC en los primeros años de vida. Las niñas con PP presentan hiperinsulinismo, dislipemia, niveles bajos de (IGFBP-1) y aumento de la grasa central. La clínica se presenta 2-3 años después de la menarquia y está precedida por un aumento de TG, LDL y de la respuesta de insulina a la sobrecarga oral de glucosa (factores predictivos de RCV y DM2). La metformina es efectiva en la prevención del hiperandrogenismo ovárico clínico en adolescentes y niñas prepuberales con historia de PP y RCIU; al mejorar el perfil endocrino-metabólico, la composición corporal y los marcadores de RCV, y evitar el hiperandrogenismo clínico; se apoya así la hipótesis de que el hiperinsulinismo es clave en la aparición del hiperandrogenismo ovárico38.
Las niñas con PP tienen la pubertad antes que la media de su población, pero la talla final suele estar en el rango familiar. Sin embargo, si hay antecedente de RCIU, la menarquia se adelanta 8-10 meses, y la talla final está como promedio una desviación estándar (aproximadamente 5 cm) debajo de lo esperado.
En niñas sin PP pero con pubertad adelantada (inicio botón mamario entre los 8 y 9 años) y antecedente de RCIU, la menarquia ocurre como promedio 1,6 años antes que en las niñas con peso adecuado para la edad gestacional, y la talla final se reduce, como término medio 5 cm39, lo que parece estar íntimamente relacionado con la RI.
Bibliografía
- Cañete R, Gil-Campos M, Aguilera CM, Gil A. Development of insulin resistence and its relation to diet in the obese child. Eur J Nutr. 2007;46:181-7.
- Gil Campos M. Relaciones entre parámetros antropométricos, ingesta de nutrientes, hormonas y lípidos plasmáticos en niños obesos. Tesis doctoral, 2004. Universidad de Córdoba.
- Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation and failure of fat cell proliferation result in ectopic fat storage, insulin resistance and type 2 diabetes mellitus. Anm NY Acad Sci. 2002;967:363-78.
- Skurk T, Alberti-Huber C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92:1023-33.
- Reaven GM. The insulin resistance syndrome: definition and dietary approaches to treatment. Annu Rev Nutr. 2005;25:391-406.
- Schaffler A, Scholmerich J, Buechler C. The role of ‘adipotropins’ and the clinical importance of a potential hypothalamic-pituitary-adipose axis. Nat Clin Pract Endocrinol Metab. 2006;2:374-83.
- Steinberg GR, Parolin ML, Heigenbauser GI, Dyck DJ. Leptin increases FA oxidation in lean but not obese human skeletal muscle. Evidence of peripheral leptin resistance. Am J Phisiol Endocrinol Metab. 2002;283:e87-92.
- Wynne K, Stanley S, McGowan B, Bloom S. Appetite control. J Endocrinol. 2005;184:291-318.
- Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54:1994-2002.
- Ahima RS. Adipose tissue as an endocrine organ. Obesity. 2006; 14 Suppl 5:s242-s245.
- Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179-83.
- Valle M, Martos R, Gascón F, Cañete R, Zafra MA, Morales R. Low grade systemic inflammation hypoadiponectinemia and high concentration of leptin are present in very young obese children and correlate with metabolic syndrome. Diabetes Metab. 2005;31:55-62.
- Stefan N, Stumvoll M. Adiponectin. Its rol in metabolism and beyond. Horm Metab Res. 2002; 43:469-74.
- Combs TP, Wagner JA, Berger J, et al. Induction of adypocite complement-related protein of 30 kilodaltons by PPAR gamma agonist: a potential mechanism of insulin sensitization. Endocrinology. 2002;143:998-1007.
- Golley RK, Magarey AM, Steinbeck KS, Baur LA, Daniels LA. Comparison of metabolic syndrome prevalence using six different definitions in overweight pre-pubertal children enrolled in a weight management study. Int J Obes (Lond). 2006;30:853-60.
- ATPIII. Executive Summary of the Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001; 285:2486-97.
- Nesto RW. The relation of insulin resistance syndrome to risk of cardiovascular disease. Rev Cardiovasc Med. 2003;4:s11-s18.
- Cook S, Weitzman M, Auinger P, Nguyen M, Dietz WH. Prevalence of a metabolic syndrome phenotype in adolescents findings from the Third National Health and Nutrition Examination Survey, 1988-1994. Arch Pediatr Adolesc Med. 2003;157: 821-7.
- Zimmet P, Alberti KG, Kaufman F, Tajima N, Silink M, Arslanian S, et al. The metabolic syndrome in children and adolescents - an IDF consensus report. Ped Diabetes. 2007;8:299-306.
- Jolliffe CJ, Janssen I. Development of age-specific adolescent metabolic syndrome criteria that are linked to the Adult Treatment Panel III and International Diabetes Federation criteria. J Am Coll Cardiol. 2007;49:891-8.
- Sinha R, Fisch G, Teague B, Tamborlane WV, Banyas B, Allen K, et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med. 2002;346:802-10.
- Atabek M, Pirgon O, Kurtoglu S. Prevalence of metabolic syndrome in obese Turkish children and adolescents. Diab Res Clin Pract. 2006;72:315-21.
- López-Capapé M, Alonso, Colino E, Mustieles C, Corbatón J, Barrio R. Frequency of the metabolic syndrome in obese Spanish pediatric population. Eur J Endocrinol. 2006;155:313-9.
- Baker J, Olsen L, Sorensen T. Childhood boy-mass index and risk of coronary heart disease in adulthood. N Engl J Med. 2007;357:2329-37.
- Goran MI, Bergman RN, Avila Q, Watkins M, Ball GDC, Shaibi GQ, et al. Impaired glucose tolerance and reduced beta-cell function in overweight latino children with a positive family history for type 2 diabetes. J Clin Endocrinol Metab. 2004; 89:207-12.
- Cruz ML, Shaibi GQ, Weigensberg MJ, Spruijt-Metz D, Ball GDC, Goran MI. Pediatric obesity and insulin resistance. Chronic disease risk and implications for treatment and prevention beyond body weight modification. Annu Rev Nutr. 2005;25:435-68.
- Franks PW, Hanson RL, Knowler WC, Moffett C, Enos G, Infante AM, et al. Childhood predictors of young-onset type 2 diabetes. Diabetes. 2007;56:2964-72.
- B Li SC, Chen W, Srinivasan S, et al. Childhood cardiovascular risk and carotid vascular changes in adulthood. The Bogalusa Heart Study. JAMA. 2003;290:2271-76.
- Beauloye V, Zech F, Tran Thi Mong H, et al. Determinants of early atherosclerosis in obeses children and adolescents. J Clin Endocrinol Metab. 2007;92:3025-32.
- Raitakari OT, Juonala M, Mahonen M, et al. Cardiovasculars risk factors in childhood and carotid artery intima-media thickness in adulthood: the cardiovascular risk study in young Finns. JAMA. 2003;290:2277-83.
- Juonala M, Vicari JSA, Mahonen M, et al. The cardiovascular risk in young Finns study. J Am Coll Cardiol. 2008;52:293-99.
- Frontini AG, Srinavasan SR, Jihua XU. Usefilness of childhood non-high density lipoprotein colesterol levels versus other lipoprotein measures in predicting adult subclinical atheroesclerosis: the Bogalusa Herat Study. Pediatrics. 2008;1098:4275 [on line].
- Lavine JE, Schwimmer JB. Nonalcoholic fatty liver disease in the pediatric population.
- Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388-93.
- Chen AK, Roberts CK, Barnard RJ. Effect of a short-term diet and exercise intervention on metabolic syndrome in overweight children. Metabolism. 2006;55:871.
- Cettour-Rose P, Samec S, Russell AP, et al. Redistribution of glucose from skeletal muscle to adipose tissue during catchup fat. A link between catch-up growth and later metabolic syndrome. Diabetes. 2005;54:751-6.
- Ibáñez L, Potau N, Ferrer A, Rodríguez-Hierro F, Marcos MV, de Zegher F. Anovulation in eumenorheic, non-obese adolescent girls born small for gestational age: insulin sensitization induces ovulation, increases lean body mass and reduce abdominal fat excess, dyslipidemia and subclinical hyperandrogenism. J Clin Endocrinol Metab. 2002;87:5702-5.
- Ibáñez L, Fucci A, Valls C, Ong K, Dunger D, de Zegher F. High eutrophil count in small-for-gestational-age children: contrasting effects metformin and growth hormone therapy. J Clin Endocrinol Metab. 2005;90:3435-9.
- Cook S. The metabolic syndrome: antecedent of adult cardiovascular disease in pediatrics. J Pediatr. 2004;145:427-30.
CONFLICTO DE INTERESES
Las autoras declaran no presentar conflicto de interés en relación con la preparación y publicación de este artículo.