
Vol. 11 - Num. 43
Grupo PrevInfad/PAPPS Infancia y Adolescencia
Cribado neonatal de metabolopatías
José Galbe Sánchez-Venturaa, Grupo PrevInfad/PAPPS Infancia y Adolescenciab
aPediatra. CS Torrero La Paz. Zaragoza. España.
bJosé María Mengual Gil (coord.).
Correspondencia: J Galbe. Correo electrónico: galbester@gmail.com
Cómo citar este artículo: Galbe Sánchez-Ventura J, Grupo PrevInfad/PAPPS Infancia y Adolescencia. Cribado neonatal de metabolopatías. Rev Pediatr Aten Primaria. 2009;11:471-84.
Publicado en Internet: 30-09-2009 - Número de visitas: 54061
Resumen
Desde finales de la década de los setenta, muchos países pusieron en marcha programas de cribado metabólico neonatal encaminados al diagnóstico precoz de aquellas metabolopatías congénitas que, detectadas en los primeros días de vida, fuesen susceptibles de tratamiento. De esta manera se mejoró notablemente el pronóstico de algunas de estas enfermedades. Inicialmente, los programas incluyeron el cribado de la fenilcetonuria y del hipotiroidismo congénito, siendo ampliados posteriormente con el de la hiperplasia suprarrenal congénita y de la fibrosis quística de páncreas. Recientemente, la introducción de la espectrometría de masas en tándem ha permitido el cribado de muchas metabolopatías, sin embargo, muchas de ellas no reúnen los criterios mínimos para ser incluidas en un programa de cribado universal.
Palabras clave
● Cribado neonatal ● Enfermedades metabólicas congénitas ● PrevenciónNota:
bGrupo PrevInfad: José Galbe Sánchez-Ventura, María Jesús Esparza Olcina, Ana Martínez Rubio, Manuel Merino Moína, Carmen Rosa Pallás Alonso, Juan Pericas Bosch, Francisco Javier Sánchez Ruiz-Cabello, Francisco Javier Soriano Faura (coordinador), Julia Colomer Revuelta, Olga Cortés Rico.
Introducción
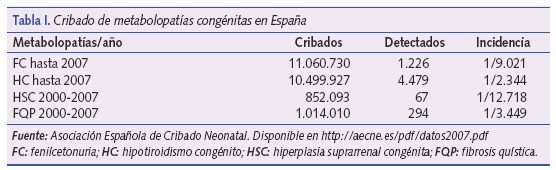
La detección sistemática de metabolopatías neonatales se inicia en España, al igual que en otros países de nuestro entorno, a finales de la década de los setenta. Desde entonces, la mayoría de los países desarrollados han introducido programas de detección de metabolopatías centrados fundamentalmente en el cribado del hipotiroidismo congénito (HC) y de la fenilcetonuria (FC). En la tabla I podemos ver el número de niños cribados en España de las distintas metabolopatías1.
Los criterios que deben cumplirse para incluir en el cribado de errores innatos del metabolismo son los siguientes2:
- Debe tratarse de una anomalía relativamente frecuente, al menos 1:15.000 recién nacidos.
- Debe producir una grave anomalía metabólica.
- Debe ser difícil de diagnosticar clínicamente en período neonatal.
- El diagnóstico clínico debe producirse tras una fase preclínica asintomática, cuando el pronóstico es malo.
- Debe existir un marcador bioquímico con una buena sensibilidad y especificidad, que permita discriminar a los recién nacidos sanos de los enfermos durante la fase preclínica.
- Debe ser posible realizar un tratamiento de la enfermedad de forma precoz, que mejore sensiblemente el pronóstico de la misma.
- El coste del programa de prevención debe ajustarse a los criterios de evaluación económica como todo programa de salud pública.
Cribado del hipotiroidismo congénito
Magnitud del problema
El hipotiroidismo fetal es causa de importantes alteraciones en el desarrollo cerebral del feto, produciendo anomalías estructurales permanentes. Las hormonas tiroideas son fundamentales en este proceso3, sobre todo para la mielinización de las fibras nerviosas y la arborización de las dentritas. La síntesis fetal de hormonas tiroideas comienza hacia la semana 10.ª-12.ª, con niveles bajos hasta la 20.ª semana. En esta fase, el feto depende de las hormonas tiroideas maternas que cruzan la placenta.
El HC rara vez se podrá diagnosticar por la sintomatología clínica en un recién nacido. Se estima que solamente un 5% de los recién nacidos con HC presentarán manifestaciones clínicas.
Prueba de cribado
Si bien lo ideal sería utilizar en todos los casos determinación de T4 y TSH, los programas de cribado adoptan una de las siguientes estrategias alternativas4-13:
- Detección primaria de T4, seguida de la de TSH en caso de que la T4 sea inferior al percentil 10.
- Determinación primaria de TSH, seguida de la de T4 cuando esta supera una cifra umbral de alrededor de 25 mUI/ml.
- Determinación de T4 y TSH simultáneamente.
- Determinación de T4 y TSH más una segunda determinación 4-6 semanas después.
En EE. UU. se ha adoptado la estrategia basada en la determinación de T4, mientras que en Europa y Japón se investiga la TSH en primer lugar. La estrategia de la T4 es más costosa desde el punto de vista económico, pero permite la detección de HC secundarios, terciarios y por déficit de TBG sin elevación de la TSH, si bien en este caso serán necesarios para el diagnóstico los test de TSH y TRF, y determinaciones de TBG. Por contra, se puede decir que estos trastornos son mucho más raros, de menor gravedad y en el caso de los HC secundarios y terciarios, generalmente asociados a otros defectos. El porcentaje de falsos positivos con la estrategia de la TSH como determinación primaria es de alrededor del 0,05%, y del 0,3% para la estrategia de la T4. Los falsos negativos suponen entre un 5% y un 10% de todos los niños con HC para cualquiera de los test de cribado9.
En la tabla II se muestra el proceso diagnóstico basado en la determinación de TSH en primer lugar3.

En las primeras 24 horas la TSH experimenta una elevación fisiológica, debido probablemente al frío, entre otros factores. Esta elevación puede dar lugar a determinaciones falsamente positivas, si se realiza el cribado en estas primeras 24 horas. Se sabe que, tras el nacimiento, los valores de T3, T4 y TSH aumentan; por causas no bien conocidas. Algunos niños prematuros, así como los recién nacidos que han precisado cuidados intensivos o han recibido tratamiento con dopamina o yodo en sus diversas formas de administración, tienen elevaciones de la TSH y amplias fluctuaciones en los niveles de T3 y T4 que impiden valorar correctamente los niveles de TSH. Los programas que se basan en dos determinaciones informan que hasta un 10% de los casos de HC se diagnostican con la segunda determinación8. Según la American Academy of Pediatrics, la prevalencia de HC para la segunda determinación es de 1:30.0004 y suelen ser niños prematuros y de muy bajo peso al nacer. Es conocido que algunas situaciones como la prematuridad, las cardiopatías o la administración de fármacos como dopamina, amiodarona o los contrastes yodados pueden causar elevaciones de la TSH. En estos casos es necesario realizar una segunda determinación pasada una semana. Casi todos los autores proponen que el cribado se realice a las 48 horas de vida4-13.
Cribado de la fenilcetonuria
La fenilcetonuria es un error innato del metabolismo en el que existe un defecto de hidroxilación de la fenilalanina (FA), que no puede convertirse en tirosina como consecuencia del déficit de fenilalanin- hidroxilasa (FAOH) o de la dihidropterina reductasa (DPHR). Esta última enzima necesita para su actuación de la presencia de tetrahidrobiopterina (BH4). La tirosina se convierte así en un aminoácido esencial para el organismo, a la vez que se produce un aumento de fenilalanina en sangre y aumenta su transaminación como vía metabólica alternativa. Se acumulan, asimismo, los ácidos fenilpirúvico, feniláctico y fenilacético. El defecto en la síntesis de FAOH se debe a una anomalía génica localizada en el cromosoma 12, y el de la DPHR en el cromosoma 4. Existen también formas con déficits parciales. La incidencia de la FC en España puede verse en la tabla I.
La FC produce un retraso psicomotor y un deterioro intelectual irreversibles en poco tiempo. Estos trastornos pueden prevenirse si se instaura precozmente una dieta pobre en fenilalanina14-20. Los niños con FC suelen ser rubios, de tez pálida y con un olor característico a paja mojada. El cribado de FC es muy eficaz, ya que antes del cribado el 85% de los niños con FC tenían un cociente intelectual (CI) menor de 40 y un 37% menor de 10. Actualmente, con el cribado, cerca del 95% de los niños con FC tienen un CI normal14,15.
Los elevados niveles de FA en sangre dan lugar a alteraciones estructurales del sistema nervioso central, con interferencia en el proceso de maduración cerebral, en la migración de los neuroblastos y en la estratificación del córtex. Hay también zonas corticales con heterotopia. Estas alteraciones neuropatológicas producen un grave retraso mental si no se inicia precozmente una alimentación pobre en FA.
El cribado de la FC se realiza mediante fluorometría, cromatografía en papel o en capa fina y últimamente mediante espectrometría de masas en tándem14-20. La clásica prueba de Guthrie ha quedado obsoleta. Para el cribado se considera adecuada la toma de muestras entre las 24 horas y los 7 días de vida (no antes de las 12 horas). No toda elevación de la FA sanguínea se debe a una FC. Existen formas debidas a inmadurez enzimática. En algunos casos la inmadurez enzimática da lugar a cifras de FA de 5 a 10 mg/dl15,20. Ver niveles de fenilalanina y actitud a seguir en la tabla III.
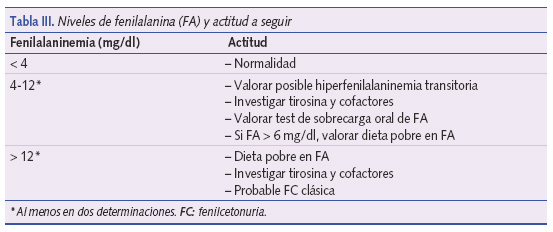
Es preciso instaurar precozmente un régimen dietético. Los niveles de FA elevados, aun durante cortos períodos de tiempo, pueden producir serios trastornos de conducta y eccema facial. Se deben monitorizar los niveles de FA, manteniéndolos entre 3 y 6 mg/dl. Hay controversia sobre el tiempo que debe durar este régimen alimentario; muchos expertos afirman que debe mantenerse unos 8-10 años. Existen, sin embargo, recomendaciones recientes en las que se insiste en prolongarlo de forma indefinida16.
Cribado de la hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (HSC) es debida, generalmente, a la ausencia o disminución de la enzima 21-hidroxilasa, que da lugar a un bloqueo en la síntesis de cortisol, con aumento secundario en la síntesis de andrógenos y virilización del feto. Se produce asimismo, con gran frecuencia, un cuadro de pérdida salina hacia las dos semanas de vida. La prevalencia en España es de alrededor de 1:15.0001. El tratamiento con hidroxicortisona estabiliza el problema y permite un crecimiento normal. En algunos casos se precisa añadir mineralcorticoides.
El cribado y la detección de portadores son posibles mediante la determinación de la 17-OH-progesterona por inmunoensayo, fluorometría o espectrometría de masas en tándem21-25. El cribado neonatal con 17-OH-progesterona tiene algunos problemas, con tasas de falsos positivos por encima del 1%. Esto se debe a que pueden darse reacciones cruzadas con otros esteroides que están elevados por estrés y también por la prematuridad9. Algunos centros de cribado proponen una estrategia con dos muestras que mejore la especificidad. Otra alternativa es la detección simultánea de 17-OH-progesterona, cortisol y androstendiona mediante espectrometría de masas en tándem, esto eliminaría la mayoría de los falsos positivos, pero plantea también algunos problemas para el cribado con muestra única21.
Cribado de la fibrosis quística de páncreas
Magnitud del problema
La fibrosis quística de páncreas (FQP) se hereda con carácter autosómico recesivo. Su frecuencia oscila entre 1:2.000 a 1:5.000 y 1 de cada 25 son portadores (tabla I). Está producida por una mutación del gen que codifica la proteína reguladora de la conductancia transmembrana o CFTR, ubicado en el brazo largo del cromosoma 7.
Se caracteriza por una anomalía exocrina generalizada, con una anormal viscosidad de todas las secreciones exocrinas que ven de esta forma dificultada su eliminación, acumulándose en los conductos excretores. En el aparato respiratorio conduce a bronconeumopatía crónica, sinusitis y poliposis nasal. En el tracto digestivo se produce en muchos casos un íleo meconial perinatal, pero también una insuficiencia pancreática con esteatorrea y desnutrición, alteraciones hepáticas con fibrosis y finalmente una verdadera cirrosis biliar. En el aparato reproductor masculino da lugar a azoospermia o agenesia de vasos deferentes. La anomalía en la CFTR lleva, asimismo, a una alteración en los canales del cloro, con una mayor concentración de NaCl en el sudor. En situaciones de sudoración profusa se pueden llegar a producir cuadros de deshidratación hiponatrémica. Es típica la colonización precoz por Pseudomona aeruginosa y Burkholderia cepacia. La enfermedad es de penetrancia variable. Desde que es posible analizar el ADN genómico se conocen más de 1.000 mutaciones para el gen de la CFTR. La más frecuente es la delta F508 (DF508), que en España representa el 50% de todas las mutaciones y el 70% en otros países.
Técnica de cribado
El primer intento de cribado para la FQP se produjo en los años cincuenta mediante el test del meconio. Desde finales de los setenta se determina la tripsina inmunorreactiva (TIR) en mancha de sangre seca, ya que la concentración plasmática de esta sustancia está aumentada en la FQP26-29. Se utiliza la inmunofluorescencia de tiempo retardado y el enzimoinmunoanálisis. Pueden considerarse normales cifras entre 90 y 200 ng/ml26. Su confirmación requiere de una segunda determinación y un test del sudor, pasado un mes. Posteriormente se ha desarrollado el análisis del ADN. Actualmente el cribado se realiza con una técnica combinada. Se utiliza la misma mancha de sangre que para otras metabolopatías. Se determina la TIR en primera instancia y si esta supera un punto de corte de 110 ng/ml, se realiza sobre la misma muestra un análisis del ADN, buscando la mutación DF508 o bien otras mutaciones. Con esta técnica secuencial TIR/ADN la sensibilidad es prácticamente del 100% y la especificidad del 99,5%.
El cribado de la FQP mejora los resultados de la enfermedad, y las poblaciones sometidas a cribado evolucionan mejor que las que se abandonan a la suerte del diagnóstico clínico26-29. El cribado permite anticipar y prevenir la deshidratación hiponatrémica por situaciones de aumento de la sudoración. El crecimiento y desarrollo de las poblaciones de FQP sometidas a cribado es mejor durante los primeros 10 años de vida, ajustando los datos para un mismo nivel de función pancreática y también es menor el deterioro de la función pulmonar. El cribado permite, asimismo, la prevención o el retraso de la colonización por Pseudomona, en muchos casos determinante de la evolución respiratoria de los niños con FQP. Algunos estudios muestran un menor número de infecciones respiratorias a los 2 años de vida en niños con FQP sometidos a cribado, respecto a los no sometidos a cribado.
Cribado de la drepanocitosis o anemia de células falciformes
Magnitud del problema
La incidencia de la anemia de células falciformes (ACF) en el colectivo subsahariano es de alrededor de 1:400. La enfermedad cursa con anemia y crisis vasooclusivas que cursan con síntomas dolorosos en cualquier punto del organismo y posibilidad de sepsis por asplenia funcional. En la ACF está indicada la profilaxis con penicilina y la vacunación antineumocócica y antimeningocócica para prevenir complicaciones infecciosas. La profilaxis con penicilina reduce hasta en un 84% la incidencia de sepsis por Streptoccoccus pneumoniae.
Técnica de cribado
El cribado se realiza mediante electroforesis de hemoglobina en sangre de cordón. Otras técnicas más sofisticadas y costosas son la cromatografía líquida o las técnicas de análisis genético. El cribado de la ACF estaría indicado en población de origen subsahariano, afroamericano o del sudeste asiático, dada la alta prevalencia en estos colectivos30,31. Debido al elevado porcentaje de población de origen subsahariano que vive actualmente en España, es conveniente que en un futuro se plantee el cribado neonatal universal de este trastorno en nuestro país.
Técnica de recogida de la muestra
Se toma una misma muestra para el cribado de HC, FC, HSC y FQP. Para el cribado de la anemia de células falciformes se utilizará sangre obtenida de cordón umbilical. Se describe a continuación la técnica para el cribado de las cuatro primeras4.
En primer lugar se desinfecta el talón con alcohol de 70º y se seca posteriormente. Se punciona con una lanceta estéril y desechable en una de las caras laterales de la parte plantar del talón. Se deja que se forme espontáneamente la primera gota de sangre, que se retira con una gasa estéril. Después se coloca el papel de filtro homologado en contacto con la segunda gota de sangre, hasta que empapa toda la superficie destinada a la mancha de sangre. La mancha debe rellenar todo el círculo dibujado en el papel y empapar bien por ambos lados, de modo que la mancha sea igual por el anverso que por el reverso. La sangre debe recogerse de una sola vez.
Los papeles se secan al aire durante 3 horas en posición horizontal, sin colocar nada encima. Deben conservarse en lugar seco y protegidos de la luz. Finalmente pueden enviarse por correo al laboratorio de referencia.
Manejo durante la punción del talón
Los cambios fisiológicos provocados por el dolor pueden producir desorganización en el desarrollo de las conexiones neuronales. Los estudios clínicos han mostrado una reducción de los cambios en los parámetros fisiológicos y en las mediciones de las puntuaciones de dolor después de la administración de analgésicos preventivos en situaciones donde el neonato presenta dolor o estrés33. Con este objetivo se han utilizado medidas no farmacológicas tales como el contacto piel con piel, caricias, amamantamiento y otro tipo de medidas de contención, como mecer o envolver con paños calientes.
La lactancia materna puede poner en marcha mecanismos endógenos de control del dolor relacionados quizás con una mayor ingesta de triptófano, elemento precursor de opioides endógenos, pero también con hechos relacionados como el contacto piel con piel con la madre, miradas, caricias, así como con la estimulación táctil, vestibular y olfatoria que se produce durante el amamantamiento33- 39. Todos los recién nacidos son capaces de experimentar dolor, incluyendo los prematuros. En una revisión sistemática de la Biblioteca Cochrane33 las medidas que se han mostrado eficaces han sido la lactancia materna durante el procedimiento, así como la administración de soluciones de glucosa o sacarosa, que se asociaron con una reducción de los cambios de la frecuencia cardíaca, la duración del llanto, el porcentaje de tiempo del llanto y de la mejoría en mediciones de dolor convalidadas y no convalidadas, cuando se compararon con placebo, con ninguna intervención o con cambio de posición. Otra revisión sistemática valoró los efectos de la sacarosa y de la succión no nutritiva. Actúan a través de los sistemas endógenos opiáceos y no opiáceos, pero los mecanismos subyacentes pueden ser diferentes38. Estos mecanismos pueden tener un efecto aditivo o sinérgico, pero es más probable que dependan del funcionamiento a nivel central. En otro ensayo clínico reciente se pudo comprobar que este efecto perduraba durante unos minutos y mejoraba el dolor y la incomodidad del niño ante procedimientos y cuidados habituales de una unidad neonatal39.
No está claro cuál es la dosis adecuada de sacarosa. Se ha propuesto entre 0,2 ml y 0,5 ml de sacarosa al 20% administrados por vía oral 2 minutos antes del pinchazo. Los efectos secundarios fueron escasos, aunque se desconocen los efectos de dosis repetidas en niños prematuros. Parece que la combinación de otras intervenciones no farmacológicas, como son el contacto visual, la estimulación táctil, el contacto piel con piel, el uso del chupete o de la succión no nutritiva, podrían potenciar los efectos beneficiosos de la sacarosa38.
Recomendaciones
- Se recomienda el cribado metabólico neonatal para HC y FC. (Fuerza de la recomendación A).
- Se recomienda el cribado para HSC y FQP. (Fuerza de la recomendación I).
- Se recomienda tomar la muestra de sangre en papel a todos los recién nacidos a partir de las 48 horas de vida lo antes posible y en cantidad suficiente para evitar falsos negativos. Igualmente en niños prematuros, enfermos graves y gemelos monocigóticos. (Fuerza de la recomendación I).
- Los niños que reciben el alta precoz en las primeras 24 horas de vida deben ser sometidos a cribado metabólico. En este caso la toma de muestra se realizará inmediatamente antes del alta. (Fuerza de la recomendación I).
- La toma de la primera muestra no debería demorarse más allá del 4.º o 5.º día de vida, ya que la detección de trastornos metabólicos y el eventual inicio de posibles tratamientos debe considerarse una urgencia. (Fuerza de la recomendación I).
- Se recomienda una segunda toma de muestra entre la 2.ª y la 4.ª semana de vida en todos los niños prematuros con peso al nacer inferior a 2.500 gramos, enfermos graves y gemelos monocigóticos. (Fuerza de la recomendación I).
- De ser posible, se debe aconsejar el amamantamiento simultáneo por su madre para aliviar el dolor en neonatos a los que se les somete a un procedimiento doloroso único, como es la punción del talón. (Fuerza de la recomendación A).
- Si no es posible el amamantamiento simultáneo, se puede administrar una solución de sacarosa como analgésico en las punciones del talón de los recién nacidos. Se propone una dosis entre 0,2 y 0,5 ml de sacarosa al 20% por vía oral, 2 minutos antes del pinchazo. (Fuerza de la recomendación A).
- La estimulación sensorial, ya sea táctil con masajes, caricias, contacto piel con piel, contacto visual, uso del chupete, envolver con paños calientes, puede reducir la respuesta dolorosa, tanto si se asocia como si no a la lactancia materna o a la administración de sacarosa. (Fuerza de la recomendación A).
- Se recomienda evitar el uso de antisépticos yodados en el período perinatal, tanto en la madre como en el niño, para evitar la aparición de hipotiroidismo transitorio en el recién nacido. (Fuerza de la recomendación B).
- Se debe comprobar en Atención Primaria que el recién nacido ha sido sometido a cribado metabólico y que existe constancia documental, tanto en la historia clínica como en el documento de salud infantil. Debe constar asimismo el resultado. (Fuerza de la recomendación I). En el caso de que en la primera revisión se compruebe o existan dudas sobre si el recién nacido ha sido sometido a cribado, se procederá a derivar al niño a la unidad de referencia o bien se realizará una determinación de forma inmediata, remitiendo en ese caso con rapidez la prueba al laboratorio de referencia. (Fuerza de la recomendación I).
Bibliografía
- Asociación Española de Cribado Neonatal [consultado el 09/08/2009]. Disponible en http://aecne.es/pdf/datos2007.pdf [actualizado el 14/ 12/2008].
- Thomason M, Lord J, Murray D, Chalmers RA, Littlejohns P, Addison M, et al. Systematic review of evidence for the appropriateness of neonatal screening programmes for inborn errors of metabolism A J Public Health. 1998;20:331-43.
- Rodríguez Sánchez MD, Pandilla Esteban ML, González Sicilia IG, Lorenzo Navarro L, Bittini Copano A, Dulín Iñíguez E, y cols. Hipotiroidismo congénito. Detección precoz. Diagnóstico y tratamiento. Acta Pediatr Esp. 1994;52:217-27.
- Alonso Fernández JR, Barreiro Conde J, Cabezón Vicente R, Castiñeiras Ramos D, Cerdá Mota T, García Caeiro AL, y cols. Guía de práctica de hipotiroidismo congénito. Avalia-t. Xunta de Galicia; 2008.
- Mayayo E, Ferrández A, Labarta J. Hipotiroidismo congénito. An Esp Ped. (Libro de actas) 1999;154-60.
- Mayayo E, Puga B, Antón R, Guallar A, Labarta JI, Ferrández A. “Screening” neonatal del hipotiroidismo congénito primario: informe del programa del Hospital Miguel Servet. Bol Soc Ar Pediatr (Zar). 1993;23(5):145-55.
- U.S. Preventive Services Task Force (USPSTF). Screening for congenital hypothyroidism. Reaffirmation Recommendation Statement March 2008 [consultado el 09/08/2009]. Disponible en www.ahrq.gov/clinic/uspstf08/conhypo/conhyprs.htm.
- American Academy of Pediatrics and American Thyroid Association. Newborn screening for congenital hypothyroidism: recommended guidelines. Pediatrics. 1987;80:745-9.
- American Thyroid Association Committee on Public Health. Screening neonatal del hipotiroidismo congénito: pautas recomendadas. Pediatrics (ed esp). 1993;35(6):346-53.
- Grüters A, Delange F, Giovanelli G, Klett M, Rochiccioli P, Torresani T. Working Group on congenital hypothyroidism of the European Society for Paediatric Endocrinology. Guidelines for neonatal screening programmes for congenital hypothyroidism. Eur J Pediatr. 1993;152:974-5.
- Screening for congenital hypothyroidism: US Preventive Services Task Force reaffirmation recommendation. Ann Fam Med. 2008;6:166.
- American Thyroid Association, Lawson Wilkins Pediatric Endocrine Society. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. 2006;117:290-303.
- Working Group of Neonatal Screening of the European Society for Pediatric Endocrinology. Revised guidelines for neonatal screening programmes for primary congenital hypothyroidism. Horm Res. 1999;52:49-52.
- Baldellou A, Tamparillas M, Salazar I. Screening of phenylketonuria. En: Proceedings of the 5° World Congress of Perinatal Medicine. Carrera J, Cabero L, Baraibar R. eds. Bologna: Monduzzi Editore; 2001.p.909-16.
- Baldellou A, Tamparillas M, Salazar I. Investigación sistemática neonatal de las hiperfenilalaninemias. Bol Soc Ar Pediatr (Zar). 1993;23(5):138-44.
- Feldman W. Screening for phenylketonuria [consultado el 09/08/2009]. Disponible en www.phac-aspc.gc.ca/publicat/clinic-clinique/pdf/s2c17e.pdf.
- Screening for phenylketonuria (PKU): US Preventive Services Task Force Reaffirmation recommendation. Ann Fam Med. 2008;6:166.
- AHRQ. Screening for phenylketonuria. March. 2008 [consultado el 09/08/2009]. Disponible en www.ahrq.gov/clinic/uspstf08/pku/pkurs.htm.
- Cocho de Juan JA, Castiñeiras Ramos DE, Fraga Bermúdez JM. Cribaje de metabolopatías. En: Sanjurjo P, Baldellou Vázquez A (eds). Diagnóstico de enfermedades metabólicas hereditarias. 2.ª ed. Madrid: Ergon; 2006.p.47-61.
- Frézal J, Farriaux JP. La phénylcetonurie hier et aujourd’hui. Bilan de l’action de dépistage néonatal systematique. Rev Prat (Paris). 1992;42(18):2316-26.
- Rey Liste T, García Caeiro AL. Neonatal screening for congenital adrenal hiperplasia: a systematic review. Endocrinol Nutr. 2007;54(4):216-24.
- Ferrández A, Mayayo E, Guallar I, Torrijo M, Sanjuan P, Labarta J. Screening neonatal para la detección de la hiperplasia suprarrenal congénita: ¿Es necesario? Bol Soc Ar Pediatr (Zar). 1993;23:180-2.
- Balsamo A, Cacciari A, Piazzi E, Cassio A, Bozza D, Pirazolli P, et al. Congenital adrenal hyperplasia: neonatal mass screening compared with clinical diagnosis only in the Emilia-Romagna Region of Italy, 1980-1995. Pediatrics. 1996;98:362-7.
- Pang S, Wallace M, Hoffmann L, Thuline H, Dorch C, Lyon I, et al. Woldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxilase deficiency. Pediatrics. 1988;81:866-74.
- Therrell B, Berenbaum S, Manter-Kapanke V, Simmank J, Prentice L, González J, et al. Results of screening 1,9 millions Texas newborn for 21- hydroxilase deficient congenital hyperplasia. Pediatrics. 1998;101:583-90.
- Paz-Valiñas L, García-Vega FJ. Cribado neonatal de la fibrosis quística. Serie Avaliación de Tecnoloxías. Informes de avaliación: INF2004/02. Santiago de Compostela: Servicio Galego de Saúde, Axencia de Avaliación de Tecnoloxías Sanitarias de Galicia, Avalia-t; 2004.
- Real Patronato sobre Discapacidad. Programas de Cribado Neonatal en España. XIV Reunión de los Centros de Cribado Neonatal 14 de octubre de 2008 [consultado el 09/08/2009]. Disponible en http://aecne.es/pdf/datos2007.pdf.
- Serra M. Cribaje neonatal de la fibrosis quística. Breus (Bar). AATM BR01;2000:1-20.
- Mérelle ME, Nagelkerke AF, Lees CM, Dezateux C. Cribaje (screening) de la enfermedad fibroquística en el recién nacido (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2008 Número 4. Oxford: Update Software Ltd. Disponible en http://www.updatesoftware. com (Traducida de The Cochrane Library, 2008 Issue 3. Chichester, UK: John Wiley & Sons, Ltd.).
- Gregg R, Simmantel A, Farrell P, Koscik R, Kosorok M, Laxova A, et al. Newborn screening for cystic fibrosis in Wisconsin: comparison of biochemical and molecular methods. Pediatrics.1997;99:819-24.
- Casado M, Barberán J, Roqueta M, Martorell Q, Bosch A, Rovira JM. Screening neonatal de drepanocitosis en el consorci Sanitari de Mataró. Justificación y primeros resultados. An Esp Pediatr. 1998;49:157-60.
- Davies SC, Cronin E, Gill M, Hickmann M, Normand C. Screening for sickle cell disease and thalassaemia: a systematic review. Health Technology Assesment. 2000;4(3):1-119.
- Pandor A, Eastham J, Beverley J, Chilcott S. Clinical effectiveness and cost-effectiveness of neonatal screening for inborn errors of metabolism using tandem mass spectrometry: a systematic review. Health Technology Assesment. 2004;8812:1-312.
- Shah PS, Aliwalas LL, Shah V. Lactancia o leche materna para los procedimientos dolorosos en neonatos (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2008 Número 4. Oxford: Update Software Ltd. Disponible en: http://www.update-software.com. (Traducida de The Cochrane Library, 2008 Issue 3. Chichester, UK: John Wiley & Sons, Ltd.).
- Association of Paediatric Anaesthetists of Great Britain and Ireland. Good practice in postoperative and procedural pain National Library of Guidelines (UK). 2008 [consultado el 09/08/ 2009]. Disponible en www.library.nhs.uk/GUIDELINESFINDER/ViewResource.aspx?resID=290188.
- Johnston C, Filion F, Campbell-Yeo M , Goulet C, Bell L, Kathryn McNaughton K, et al. Kangaroo mother care diminishes pain from heel lance in very preterm neonates: a crossover trial C. BMC Pediatrics. 2008;8:13. Doi:10.1186/1471-2431-8-13.
- Ludington-Hoe SM, Hossein RB. Skin-toskin contact analgesia for preterm infant heel stick. AACN Clin Issues. 2005;16(3):373-87.
- Codipietro L, Ceccarelli M, Ponzone A. Breastfeeding or oral sucrose solution in term neonates receiving heel lance: a randomized, controlled trial. Pediatrics. 2008;122;e716-21. DOI: 10.1542/peds.2008-0221.
- Yamada SB, Ohlsson A. Sucrosa para la analgesia en recién nacidos sometidos a procedimientos dolorosos (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2008 Número 4. Oxford: Update Software Ltd. Disponible en http://www.update-software.com (Traducida de The Cochrane Library, 2008 Issue 3. Chichester, UK: John Wiley & Sons, Ltd.).
- Taddio A, Shah V, Katz J. Reduced infant response to a routine care procedure after sucrose analgesia. Pediatrics. 2009;123;e425-9. DOI: 10.1542/peds.2008-3028.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.