
Vol. 25 - Num. 98
Original Papers
Evaluation and follow up of patients with short stature in Primary Care
Elena García-Ochoa Fernándeza, M.ª Elena Cabezas Tapiab, Mireya Orío Hernándezc
aServicio de Pediatría. Hospital Universitario Rey Juan Carlos. Móstoles. Madrid. España.
bPediatra. CS de Villaviciosa de Odón. Villaviciosa de Odón. Villaviciosa de Odón. Madrid. España.
cPediatra. CS Alcalde Bartolomé González. Móstoles. Madrid. España.
Correspondence: ME Cabezas. E-mail: cabezastapia@hotmail.com
Reference of this article: García-Ochoa Fernández E, Cabezas Tapia ME, Orío Hernández M. Evaluation and follow up of patients with short stature in Primary Care . Rev Pediatr Aten Primaria. 2023;25:155-63.
Published in Internet: 09-05-2023 - Visits: 18216
Abstract
Introduction: anthropometric assessment is one of the functions of the Primary Care pediatrician. It is important to include it in healthy children check-ups in order to detect those cases of short stature, study them, refer them to Endocrinology if necessary and carry out the corresponding treatment.
Objective: to describe and analyze the management of children with short stature in a primary care health center.
Material and methods: retrospective descriptive study on children with short stature between 2 and 16 years of age, whose Health Center is Villaviciosa de Odón, from January 1, 2020 to January 1, 2022. The medical records were selected in the Consult@web application and reviewed in AP Madrid and Horus. Statistical analysis was performed using SPSS.
Results: 62 patients were studied, of whom 19 met the criteria for short stature. 16% had a family history of short stature and 16% had a history of constitutional delay of growth. Mean height at diagnosis was -2,36 ± 0,49 SD. The most frequently requested complementary tests were bone age (74%) and blood tests (78%). The most frequent diagnosis was idiopathic short stature (58%). 32% received growth hormone treatment. Follow-up was carried out exclusively in primary care in 32%. Given the small sample size, no statistical significance was obtained in the comparisons.
Conclusions: short stature is a frequent reason for consultation in Primary Care, being important to perform a complete evaluation recognizing those alarming data that lead to suspect associated pathology.
Keywords
● Constitutional delay of growth ● Familiar short stature ● Growth ● Idiophatic short statureINTRODUCTION
Growth is a complex biological process determined by genetic, hormonal and environmental factors,1,2 as well as an excellent indicator of general health.1
Short stature (SS) is a frequent reason for consultation at the primary care (PC) level.3,4 It is defined as a height z score of less than −2 or below the 3rd percentile for age, sex and race, a short stature more than 2 standard deviations (SDs) below the target height3-5 or a height velocity z score of less than −1 for 2 or 3 years.3,4
Short stature is classified based on its aetiology as:
- Idiopathic short stature (ISS): no evidence of disease, with normal anthropometric measurements at birth, without dysmorphic features and with adequate secretion of growth hormone (GH).4,6 This includes the following normal variants: familial short stature (FSS) and constitutional delay in growth and puberty (CDGP), which account for approximately 80% of total cases.7,8
- Short stature with pathological aetiology: secondary to tumours, prolonged treatment with glucocorticoids, GH deficiency, genetic syndromes or systemic diseases such as chronic kidney disease, congenital heart disease, coeliac disease, inflammatory bowel disease, immunodeficiency, metabolic disorders, rickets, hypothyroidism or hypercortisolism.5,8
The diagnosis of SS is made by the PC paediatrician in relation to the reference population and requires differentiating pathological processes requiring treatment from normal variants of SS.6-8
The cornerstone of diagnosis is the history-taking and physical examination.2,7,8 The anamnesis must cover the prenatal, perinatal and family history, with particular emphasis on the presence of genetic disorders, pubertal development and the height of both parents, from which the target or genetic height is calculated applying the Tanner formula8 (the expected final height of the child of a given couple, assuming normal inheritance). The examination must include a thorough anthropometric evaluation, assessing whether the SS is harmonic through the measurement of body segments and arm span.3,5,8 Attention must also be paid to sexual development, dysmorphic features and skin abnormalities.3,5,8
Another tool that can be used in the assessment is bone age, determined through comparison with the Greulich and Pyle atlas,9 which allows prediction of the final height that would be achieved if the patient remained in the current percentile, and calculation of the predicted height through models such as the Bayley-Pinneau method.6,8
Few patients require a more detailed evaluation to establish the aetiology,3,8 including blood tests and karyotyping; so it is important to manage these aspects from the PC level.5,7
The objective of the study was to describe and analyse the management of SS at the PC level to identify opportunities of improvement in everyday clinical practice.
MATERIAL AND METHODS
We conducted a retrospective, observational and descriptive study by reviewing the electronic health records in the PC database of the public health system of Madrid (AP Madrid), with the prior authorization of the Research Ethics Committee of the Fundación Jiménez Díaz (Madrid). Since the study was not experimental, it was exempted from informed consent. We safeguarded the anonymity of the patients throughout data collection and the study.
We included children in the catchment population of the primary care centre of Villaviciosa de Odón (Madrid) with a diagnosis of SS between January 1, 2000 and January 1, 2022, who were aged 2 to 16 years at the time of diagnosis. To select the patients, we reviewed electronic health records identified by means of Consult@web, the International Classification of Primary Care and a free search.
Inclusion criteria
- Patient with SS, defined as: height for age and sex more than 2 SDs below the mean using the Hernandez 1988 tables as reference10; height more than 2 SDs below the target height; height velocity z score of less than −1 for at least 2 years.
- Age 2-16 years at diagnosis.
- History of small for gestational age (SGA) and absence of catchup growth at age 2 years.
- Diagnostic code or free text in health record documenting: SS, CDGP, SGA, intrauterine growth restriction (IUGR) or growth delay (T10 code).
We reviewed the health records of the patients to collect the variables presented in Table 1.
| Table 1. Assessment and followup of patients with short stature at the primary care level. Study variables | |
|---|---|
| Qualitative variables | Quantitative variables |
| Nominal/Dichotomous | Continuous |
| Sex | Birth length (cm) |
| Intrauterine growth restriction | Birth length (percentile) |
| Small for gestational age | Birth length (z) |
| Breastfeeding | Birth weight (cm) |
| Preterm birth | Birth weight (percentile) |
| Feeding/eating problems | Birth weight (z) |
| Chronic medication | Birth head circumference (cm) |
| Type of chronic medication | Birth head circumference (percentile) |
| Underlying disease | Birth head circumference (z) |
| Type of underlying disease | Maternal height (cm) |
| Family history of CDGP | Maternal height (percentile) |
| Family history of SS | Paternal height (cm) |
| Dysmorphic features | Paternal height (percentile) |
| Bone age study (yes or not) | Target height (cm) |
| Abnormal test result | Target height (percentile) |
| Other diagnostic tests | Target height (z) |
| Type of diagnostic tests | Height at diagnosis (cm) |
| Treatment | Height at diagnosis (z) |
| Type of treatment | Weight at diagnosis (kg) |
| Followup | Weight at diagnosis (z) |
| Bone age (advanced, delayed, appropriate) | Body mass index (kg/m2 and z) |
| Blood tests (normal, abnormal, not performed) | Height velocity (cm and z) |
| Final diagnosis | Predicted adult height (cm) |
| Ordinal | Discrete |
| Tanner stage at diagnosis | Age at diagnosis |
| Duration of breastfeeding | |
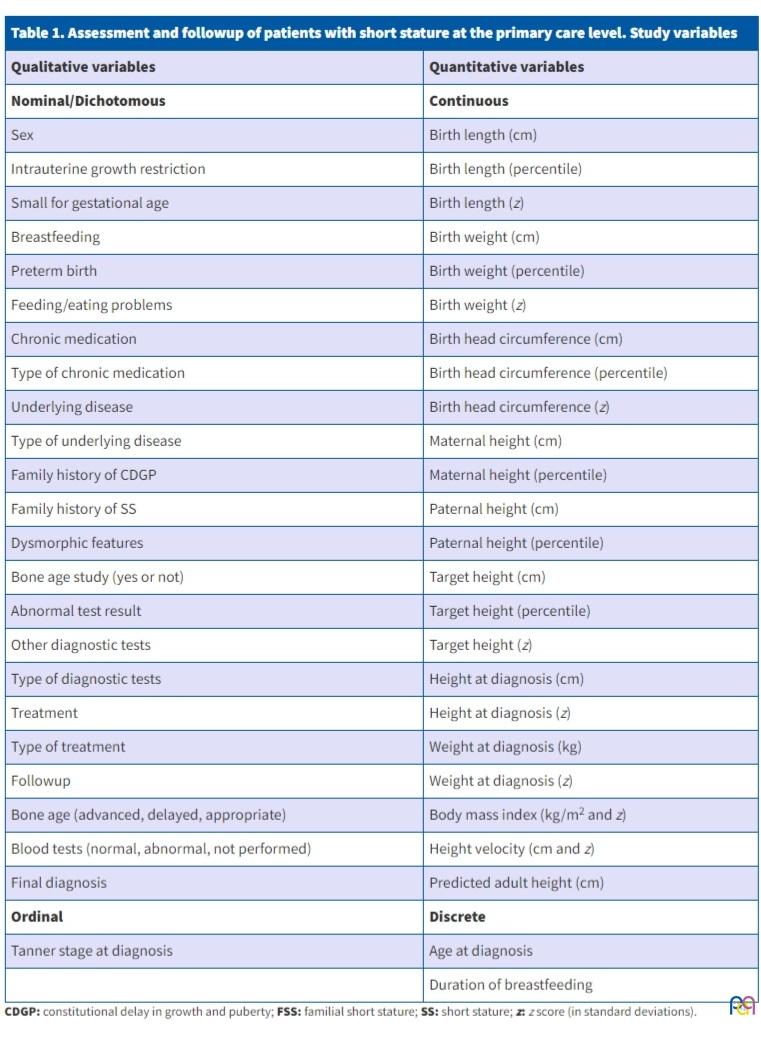
For the anthropometric parameters, we used the 1988 growth tables of Hernandez,10 and we calculated the height z score and the predicted height with the software tool provided at www.webpediatrica.com.
The statistical analysis was performed with the software package IBM SPSS version 22 for Windows.
Statistical analysis
First we assessed whether the data followed a normal distribution by means of the Kolmogorov-Smirnov test.
We summarised qualitative variables as frequencies and quantitative variables as mean ± SD or median and interquartile range. We compared normally distributed continuous variables with the Student t test and variables with a nonparametric distribution with the Mann-Whitney U test. we assessed the association between variables by means of the Spearman rank correlation coefficient.
We considered results statistically significant if the p value was less than 0.05, which corresponds to a 95% level of confidence.
RESULTS
We identified 62 candidates. Of this total, only 19 (30.6%) met the inclusion criteria. Of the 43 excluded patients, 23 (54%) sought care for SS, but did not meet the anthropometric criteria; 6 (14%) only had slow weight gain; 5 (12%) delayed puberty; 4 (9%) had been diagnosed in infancy; 4 (9%) had precocious puberty with a low predicted height, but did not meet the criteria for SS; y 1 (2%) were followed up in a private facility, so that further data could not be obtained.
Of the included patients, 11 were male (58%) and 8 female (42%). The mean age was 7 years, and the median 8 years (2-14), with an interquartile range of 8 years (3-10).
As regards the prenatal history, 2 (10.5%) had a diagnosis of IUGR, while 15 (79%) had normal prenatal ultrasounds. This information was not available in the remaining 2 (10.5%).
The anthropometric measurements at birth were normal in 68% (13), while 10.5% (2) met the criteria for SGA. In 21% (4), they had not been documented in the health record. Using the available data, we found the following:
- The mean birth length was 48 ± 2.3 cm (41.5-51). The mean z score of the length for gestational age and sex was −1 ± 0.8 (−2.1 to −0.1).
- The mean birth weight was 3093 ± 538 g (1730-3795). The mean birth weight z score was −0.2 ± 1 (−1.6 to 1.4).
Only 1 patient (5%) was born preterm, with anthropometric measurements at birth in the normal range for gestational age.
As regards nutrition, 52.6% were breastfed (10) and 15.8% formula-fed (3), while the feeding modality was not documented in the records of 31.6% (6). There was a history of feeding problems in 37% (7) (breastfeeding problems, gastro-oesophageal reflux or picky eater).
Comorbidities were documented in 42% (8): 3 had attention-deficit hyperactivity disorder (ADHD); 1 global developmental delay; 1 severe atopic dermatitis and childhood eating disorder; 1 Noonan syndrome; 1 pneumococcal meningitis and chronic kidney disease (CKD) grade III as a complication; and 1 autism spectrum disorder (ASD). Of these patients, 50% was taking medication: the patient with meningitis and CKD received antiepileptic drugs, calcium and bicarbonate; the patient with severe atopic dermatitis, dupilumab; 2 of the patients with ADHD, methylphenidate.
The health records documented FSS in 16% (3), and parental height was only documented in the records of 2 of these patients, with a maternal height at or below the 3rd percentile and a normal paternal height.
Parental heights had only been documented in the records of 15 out of the 19 patients under study:
- The mean maternal height was 157.1 ± 6 cm (147-168), corresponding to the 24th percentile (z score: −0.7) in the 1988 Hernández growth charts.
- The mean maternal height was 175.1 ± 7.1 cm (165-192), corresponding to the 47th percentile (z score: −0.1).
On the other hand, 3 patients (16%) had a family history of CDGP, in every case in the mother. IN 9 patients (47%), the pubertal development of the parents had been normal, while in 7 (37%) this aspect had not been documented.
Off the 19 patients under study, 4 had dysmorphic features (21%): one was the patient with Noonan syndrome, one the patient with global developmental delay and in the other two this fact was documented without further information.
In terms of pubertal development, at the time of diagnosis 58% (11) were at Tanner stage I, 11% (2) at stage II and 5% (1) at stage V. This datum had not been documented in 26% (5).
The anthropometric values at diagnosis were:
- The mean z score of the weight for age and sex was −1.2 ± 0.7 (data for 18 patients, no data available for the patient followed up in the private system).
- The mean z score of the weight for age and sex was −2.4 ± 0.5 (data available for all 19 patients).
We were able to calculate the height velocity in 15 patients (79%), and found a mean z score of 0.7 ± 1.7. It was only below the 3rd percentile in 3 patients, two of who had GH deficiency and one ISS.
A bone age study was performed in 14 patients (74%), and bone age was found to be appropriate for age in 5 (36%), advanced in 1 (7%) and delayed in 7 (50%). The result had not been documented in 1 patient (7%). A bone age report was only available at the same time the height was measured in 8 of the patients, making it possible to calculate the predicted adult height with the Bayley-Pinneau method, with a mean value of 161.5 cm (z score: −2.3) in male patients (6) and 145.5 cm (height: z −2.8) in female patients (2).
Blood tests were ordered in 15 patients (79%) and considered unnecessary in the rest. The results were normal in 9 patients (60%), while abnormalities were detected in 6 (40%) (hypercholesterolemia in 2, low levels of insulin-like growth factor 1 [IGF1] in 3 and findings compatible with CKD in 1). Testing for IGF1 is not available at the PC level, and these values were obtained by reviewing hospital records.
Ten patients (53%) underwent additional diagnostic tests at the hospital level, including: clonidine challenge test, insulin tolerance test, karyotyping and other genetic tests, and a brain magnetic resonance imaging scan.
Eleven patients received a diagnosis of ISS (58%), in 4 cases of FSS and in 1 of CDGP. Of the remaining patients, 3 received a diagnosis of GH deficiency (16%); 1 of Noonan syndrome (5%); 1 of SGA without catchup growth (5%); 1 of SS in the context of CKD (5%), and 2 who had received the diagnosis of short stature recently were still under evaluation to determine the definitive diagnosis (11%).
Thirty-two percent of the patients (6) received GH: 3 due to GH deficiency, 1 due to CKD, 1 due to SGA without catchup growth; and 1 for unknown reasons, as the followup took place in a private health care facility.
Thirty-two percent of patients (6) were followed up exclusively at the PC level: 2 with FSS, 2 with a recent diagnosis undergoing evaluation, 1 with CDGP and 1 with ISS. The remaining 68% (13) required evaluation at the hospital level.
Although the study followup was long, the sample was small, so we did not obtain statistically significant results for the associations under study.
Table 2 presents the comparison of qualitative study variables and the mean height z score at diagnosis, performed by means of the Mann-Whitney U test, with the corresponding p values.
| Table 2. Assessment and followup of patients with short stature at the primary care level. Association of qualitative variables with the height at diagnosis (z score) |
|||
|---|---|---|---|
| Variable | Mean height at diagnosis (z score) | Statistical significance(p <0.05) | |
| History of CDGP | Yes | -2.17 | 0.35 |
| No | -2.23 | ||
| History of FSS | Yes | -2.20 | 1.00 |
| No | -2.23 | ||
| Sex | Male | -2.39 | 0.97 |
| Female | -2.32 | ||
| CIR | Yes | -3.11 | 0.41 |
| No | -2.28 | ||
| PEG | Yes | -3.11 | 0.44 |
| No | -2.27 | ||
| Preterm birth | Yes | -2.30 | 0.86 |
| No | -2.36 | ||
| Breastfeeding | Yes | -2.48 | 0.87 |
| No | -2.33 | ||
| Feeding problems | Yes | -2.48 | 0.67 |
| No | -2.29 | ||
| Dysmorphic features | Yes | -2.22 | 0.69 |
| No | -2.40 | ||
| Underlying disease | Yes | -2.14 | 0.09 |
| No | -2.49 | ||
| Previous medication | Yes | -2.10 | 0.15 |
| No | -2.43 | ||
| Bone age study performed | Yes | -2.27 | 0.87 |
| No | -2.33 | ||
| Other diagnostic tests | Yes | -2.08 | 0.79 |
| No | -2.45 | ||
| Followup | AP | -2.22 | 0.38 |
| Hospital | -2.43 | ||
| Treatment | Yes | -2.56 | 0.93 |
| No | -2.27 | ||
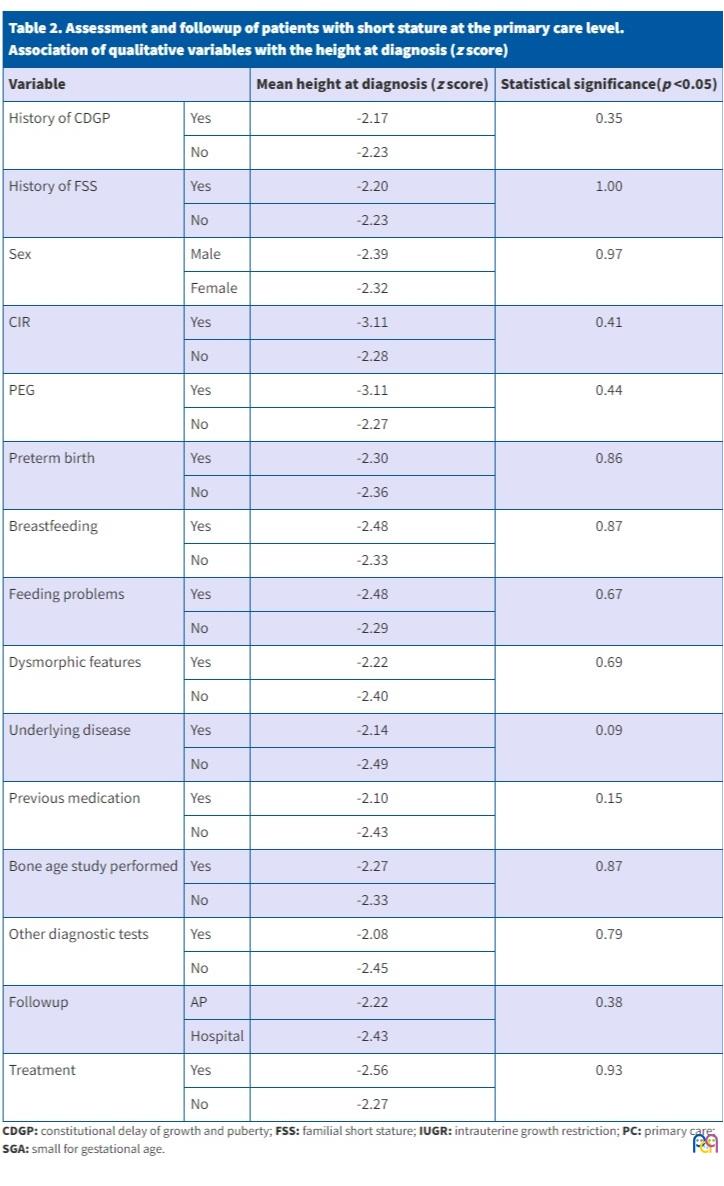
Table 3 presents the comparison of qualitative study variables and the mean age at diagnosis, performed by means of the Student t test, with the corresponding p values.
| Table 3. Assessment and followup of patients with short stature at the primary care level. Association of qualitative variables with the age at diagnosis |
|||
|---|---|---|---|
| Variable | Mean age at diagnosis | Statistical significance(p <0.05) | |
| History of CDGP | Yes | 4 | 0.054 |
| No | 6.7 | ||
| History of FSS | Yes | 11.33 | 0.41 |
| No | 5.69 | ||
| IUGR | Yes | 6 | 0.75 |
| No | 7.53 | ||
| SGA | Yes | 6 | 0.69 |
| No | 7.08 | ||
| Breastfeeding | Yes | 5.4 | 0.24 |
| No | 7.67 | ||
| Feeding problems | Yes | 5.57 | 0.64 |
| No | 7.83 | ||
| Underlying disease | Yes | 7.86 | 0.17 |
| No | 6.5 | ||
| Previous medication | Yes | 9.5 | 0.34 |
| No | 6.33 | ||
| Followup | PC | 8.25 | 0.55 |
| Hospital | 6.09 | ||
| Treatment | Yes | 7.5 | 0.91 |
| No | 6.77 | ||
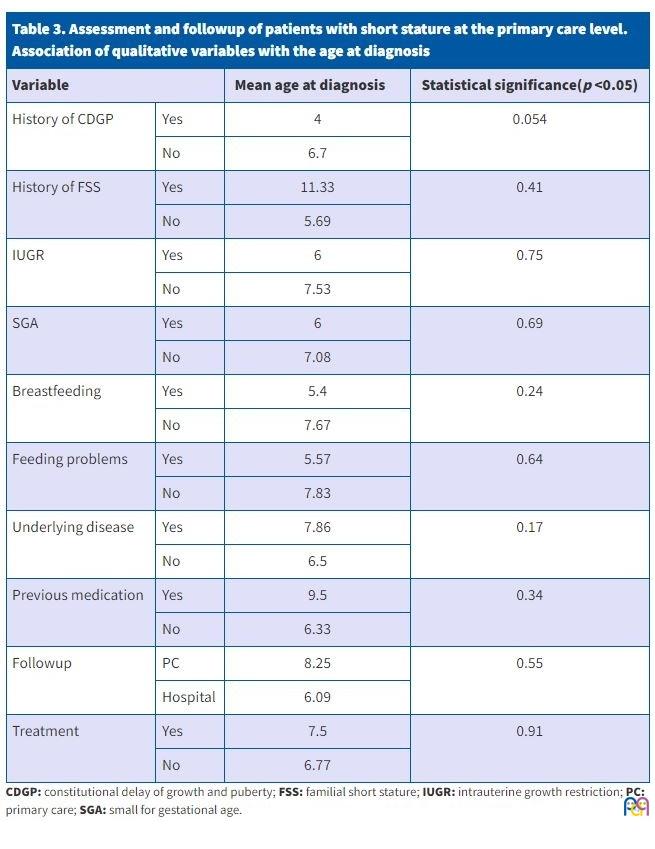
The diagnosis of SS was made earlier in patients with a family history of CDGP, with a p value that neared the threshold of statistical significance (p = 0.054).
Table 4 presents the correlations between the quantitative variables and the height z score at diagnosis, assessed by means of the Spearman r, with the corresponding p values.
| Table 4. Assessment and followup of patients with short stature at the primary care level. Association of quantitative variables with the height at diagnosis (z score) |
|
|---|---|
| Variable | Statistical significance (p <0.05) |
| Age | 0.15 |
| Breastfeeding duration | 0.76 |
| Birth length (cm) | 0.80 |
| Birth length (z) | 0.80 |
| Birth weight (g) | 0.83 |
| Birth weight (z) | 0.15 |
| Predicted height | 0.09 |
| Maternal height (cm) | 0.71 |
| Paternal height (cm) | 0.29 |
| Target height (z) | 0.48 |
| Height velocity (z) | 0.35 |
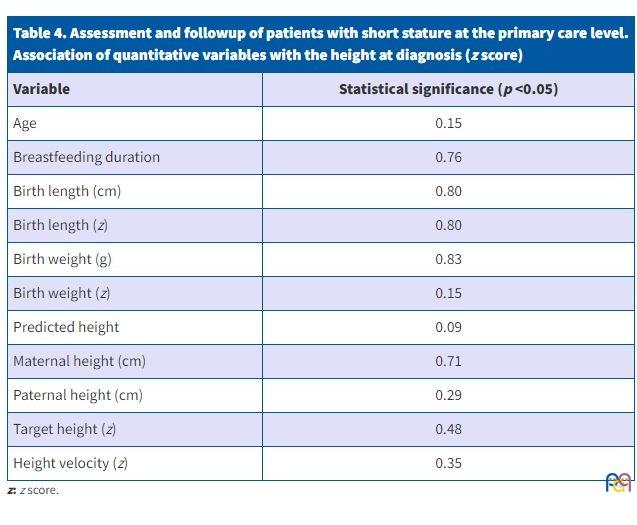
DISCUSSION
The number of visits for SS is increasing in relation to the social perception of height as a health indicator and current beauty standards.11 In many cases, a thorough history-taking and physical examination and the performance of first-line diagnostic tests can guide the diagnosis without requiring referral to hospital-based care in many cases.
Growth assessment is included in well-child programmes, including the guidelines of the PrevInfad Child and Adolescent Prevention group of the Asociación Española de Pediatría de Atención Primaria (Spanish Association of Primary Care Paediatrics, AEPap), for every paediatric age group.12
In consequence, monitoring growth in the framework of well-child preventive care programmes should be part of the routine tasks of PC paediatricians. A child who exhibits adequate growth is not likely to have significant disease.5,11
In Spain, longitudinal growth studies have been conducted at different times, and there have been discrepancies between them and in comparison to the study conducted by the World Health Organization (WHO), as they all had different inclusion criteria. The WHO study included patients from different countries and took into account nutritional parameters, and use of the WHO standards is recommended in young children and infants, especially those who are breastfed, as there is no evidence of differences based on racial or ethnic background. After this period, the use of references based on the compilation of data from studies conducted in Spain is considered more appropriate.13 In the case of certain diagnoses, such as Turner, Noonan or Down syndrome, specific charts are used for reference.13
In the PC setting, the WHO growth standards are the most widely used reference, while the growth charts of Hernández of 198810 or of the Spanish Growth Study of 2010 are used more frequently at the hospital level. For the reasons noted above, and since the sample under study spanned a long period starting in year 2000, we chose to use the Hernández growth charts of 1988 as reference.10
In regard to the findings of the study, it is worth noting that of all the patients whose health records had documentation of SS through a diagnostic code or free text, 70% did not meet the criteria for SS, a percentage that was higher compared to other studies, in which it is approximately 50%.7 Some of the possible reasons for this are worry by the family about the height of the patient, due to how height is perceived in society today, as we already noted, incorrect height measurement, the use of inappropriate growth references or incorrect coding, as the code that is entered first is often the presenting complaint as opposed to the actual diagnosis.
As for the family history, it is important to take into account that FSS frequently goes undetected, as SS may be perceived as normal within the family in this context.8 Thus, parental heights should be documented in the first PC visit to ensure that patients with pathological SS who also have a history of FSS are identified.
In relation to the physical examination, we ought to highlight that the anthropometric assessment of body segment proportions and arm span was not performed or recorded in any of the patients.
A salient finding in our series was the high proportion of patients with dysmorphic features (21%), which may be related to the greater frequency of health care visits by patients with pre-existing diagnoses.
Idiopathic short stature is a diagnosis of exclusion, and it can only be confirmed by the evolution of the patient’s growth and performance of diagnostic tests as indicatrd.6,14 In our sample, a precise diagnosis was achieved in half of the patients, in agreement with the previous literature.4 The most frequent diagnosis was ISS (58%), followed by GH deficiency, in contrast to studies conducted in the hospital setting in which, while the most frequent diagnosis is also ISS, the next most frequent diagnosis is SS associated with SGA.1
As regards the high frequency of ISS, results in the previous literature have been heterogeneous,1,7,15 due in part to the gradual discovery of causes of SS in cases previously categorised as idiopathic, and differences between centres in the availability of diagnostic methods, for instance genetic testing.1
Another salient finding was the high proportion of GH deficiency cases (16%), greater compared to the studies published by Sevilla Ramos et al. (4%)1 and of Rodríguez et al. (5.1%),7 although similar to the proportion found by Majcher et al. (18.7%).15 Treatment with GH was also more frequent than reported in other studies (32% versus 15%).1
As regards the limitations of the study, we ought to highlight its retrospective design, based on the review of health records, and the fact that many PC health records were missing key information necessary for the assessment of a patient with SS. This made it necessary to review hospital records to complete the data collection.
In addition, the sample was small and came from a specific population, which affects the validity and magnitude of the results as well as their generalizability, and none of the associations under study met the established threshold for statistical significance.
We found that multiple studies on SS have been conducted in the hospital setting, but very few in the PC setting. In light of all of the above, it would be useful to conduct a multicentre study with participation of other primary care centres and other health care areas to be able to draw significant conclusions.
CONCLUSION
The PC paediatrician is usually the provider in charge of assessing height and height velocity, and therefore is the first to detect abnormalities in height, both as the presenting complaint in on-demand visits or in the context of routine checkups in the context of the well-child programme.
In the sample under study, we found errors in diagnosis, coding and documentation in the health record, which did not meet current standards. Thus, it would be interesting to assess the approach to the management of patients with SS at the PC level in other centres and catchment areas by means of multicentre studies to determine whether these errors are common, identify the causes and implement the necessary measures to optimize the use of resources and the management of patients with SS.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare in relation to the preparation and publication of this article.
This study did not receive any form of external funding. It was presented in June 2022 as a master’s dissertation in the Master’s Programme in Primary Care Paediatrics of the Universidad Complutense de Madrid.
AUTHORSHIP
Author contributions: principal investigator (EGF), collaboration and revision (MECT, MHO).
ABBREVIATIONS
ADHD: attention-deficit hyperactivity disorder· ASD: autism spectrum disorder · CDGP: constitutional delay in growth and puberty · CKD: chronic kidney disease · FSS: familial short stature · GH: growth hormone · IGF1: insulin-like growth factor-1 · ISS: idiopathic short stature · IUGR: intrauterine growth restriction · PC: Primary Care · SD: standard deviation · SGA: small for gestational age · SS: short stature · WHO: World Health Organization.
REFERENCES
- Sevilla Ramos MP, Alija Merillas MJ, López Andrés N. Diagnóstico de Talla baja en consulta de endocrinología pediátrica de un hospital provincial secundario. Rev Esp Endocrinol Pediatr 2014;5:9-17.
- Zhou E, Hauser BR, Hee Jee Y. Genetic evaluation in children with short stature. Curr Opin Pediatr 2021;33:458-63.
- De Paz Marín S. Qué hacer ante un niño con talla baja. Form Act Pediatr Aten Prim. 2009;2:89-94.
- Peix Sambola MA, Riaño Galán I. Guía de Algoritmos en Pediatría de Atención Primaria. Talla baja. AEPap 2019 [online] [accessed 25/04/2023]. Available at https://algoritmos.aepap.org/algoritmo/64/talla-baja
- Ibáñez Toda l, Marcos Salas MV. Abordaje de la talla baja. AEPap ed. Curso Pediatría 2015. Madrid: Lúa Ediciones 3.0; 2015. p. 85-94 [online] [accessed 25/04/2023]. Available at www.aepap.org/sites/default/files/cursoaepap2015p85-94.pdf
- Bogarín R, Richmond E, Rogol AD. A new approach to the diagnosis of short stature. Minerva Pediatr. 2020;72:250-62.
- Rodríguez M, Lang R, Lucas NJ, Méndez V. Estudio prospectivo de un grupo de niños con talla baja o disminución de la velocidad de crecimiento, o ambas. Rev Med Uruguay. 2005;21:49-58.
- Pozo Román J. Talla baja idiopática y variantes normales de talla baja. Pediatr Integral. 2020;XXIV:208-21.
- Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand and wrist. 2nd Ed. Stanford: Stanford University Press; 1959.
- Hernández M, Castellet J, Narvaíza JL, Rincón JM, Ruiz I, Sánchez E, et al. Curvas y tablas de crecimiento. Madrid: Ediciones Garsi; 1988.
- de Onis M, Branca F. Childhood stuntig: a global perspective. Mater Child Nutr. 2016;12:12-26.
- Grupo PrevInfad/PAPPS Infancia y Adolescencia. Guía de actividades preventivas por grupos de edad. In: PrevInfad / PAPPS [online] [accessed 25/04/2023]. Available at https://previnfad.aepap.org/recomendacion/actividades-por-edad-rec
- Carrascosa Lezcano A, Fernández García JM, Ferrández Longás A, López de Lara D, López Siguero JP. Estudios españoles de crecimiento: Situación actual, utilidad y recomendaciones de uso. An Pediatr (Barc). 2011;74:193.e1-193.e16.
- López Siguero JP, Ariza Jiménez AB. Talla baja de etiología no determinada y cada vez menos idiopática. Rev Esp Endocrinol Pediatr. 2021;12(1):21-34.
- Majcher A, Bielecka Jasiocha J, Pyrzak B. Analysis of reasons of short stature in own material. Pediatr Endocrinol Diabetes Metab. 2009;15:152-6.