

Vol. 23 - Num. 92
Casos clínicos en Digestivo
Quiste de colédoco: nunca bajar la guardia
Marta García Vegaa, Sandra Pérez Salasa, Iván Carabaño Aguadoa, Elisa Aguirre Pascualb, Indalecio Cano Novilloc, María López Díazc
aSección de Gastroenterología, Hepatología y Nutrición Pediátrica. Servicio de Pediatría. Hospital Universitario 12 de Octubre. Madrid. España.
bSección de Radiología Infantil. Servicio de Radiodiagnóstico. Hospital Universitario 12 de Octubre. Madrid. España.
cServicio de Cirugía Pediátrica. Hospital Universitario 12 de Octubre. Madrid. España.
Correspondencia: M García. Correo electrónico: mgarciavega6@gmail.com
Cómo citar este artículo: García Vega M, Pérez Salas S, Carabaño Aguado I, Aguirre Pascual E, Cano Novillo I, López Díaz M. Quiste de colédoco: nunca bajar la guardia. Rev Pediatr Aten Primaria. 2021;23:425-8.
Publicado en Internet: 16-12-2021 - Número de visitas: 10989
Resumen
El quiste del colédoco es una dilatación congénita de la vía biliar. La presentación clínica es inespecífica y a menudo supone un dilema diagnóstico. Se debe tener un elevado índice de sospecha ante casos de ictericia, dolor abdominal y masa abdominal palpable. Para su diagnóstico es fundamental un estudio de imagen, siendo el indicado de forma inicial la ecografía. Pueden presentar múltiples complicaciones, incluyendo colangitis, pancreatitis, colangiocarcinoma, cirrosis biliar y hepática. Para su manejo se recomienda la resección del quiste, para evitar complicaciones y disminuir el riesgo de malignización.
Se recomienda un seguimiento a largo plazo, con ecografías y análisis anuales, dado que el riesgo de malignización se mantiene elevado incluso tras la escisión del quiste. El abordaje óptimo de esta patología requiere un enfoque multidisciplinar, desde la sospecha diagnóstica que a menudo procederá de los servicios de Atención Primaria, incluyendo también gastroenterólogos, cirujanos, patólogos y radiólogos.
Palabras clave
● Colédoco ● Colestasis ● Ictericia ● Quistes ● Vía biliarINTRODUCCIÓN
Se presenta el caso de una lactante de tres meses diagnosticada de quiste de colédoco, una entidad rara cuya presentación clínica suele ser inespecífica. Para su diagnóstico se necesita un alto índice de sospecha, pudiendo diagnosticarse a cualquier edad o incluso como hallazgo incidental. Un diagnóstico precoz y un manejo adecuado son fundamentales para obtener un resultado óptimo y con buen pronóstico.
CASO CLÍNICO
Lactante mujer de tres meses, sin antecedentes de interés, remitida a Urgencias desde Atención Primaria por cuadro de ictericia de dos semanas de evolución. Alimentada con lactancia mixta, con adecuada tolerancia. Afebril. No refieren coluria ni acolia. Adecuada ganancia ponderoestatural. No muestra irritabilidad ni otros síntomas asociados.
En la exploración física destaca ictericia mucocutánea generalizada, con abdomen levemente distendido, pero blando y depresible, no doloroso, y hepatomegalia de tres traveses, no otras masas ni visceromegalias.
Durante su estancia en observación se objetivan deposiciones acólicas. En el análisis de sangre destaca un perfil hepático alterado (colestasis con hipertransaminasemia), con AST 280 U/l, ALT 345 U/l, GGT 1534 U/l, FA 880 U/l, LDH 450 U/l, bilirrubina total 8,42 mg/dl (directa 8,11), procalcitonina 0,38 ng/ml, proteína C reactiva 3,8 mg/l. Coagulación normal.
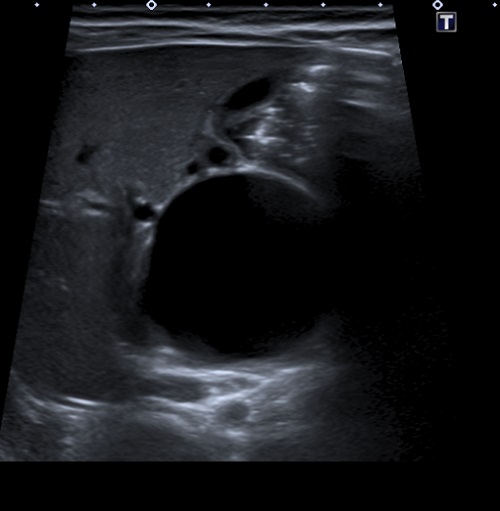
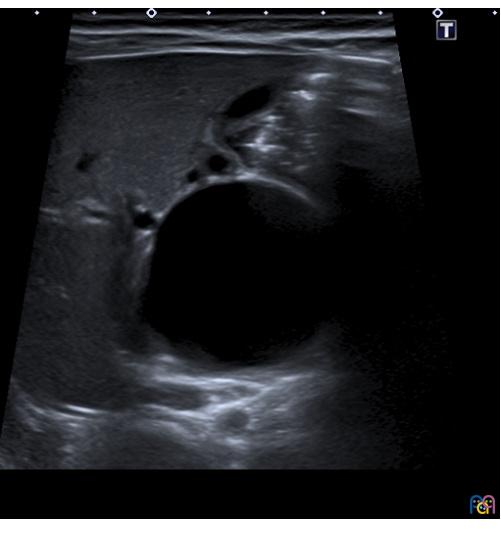
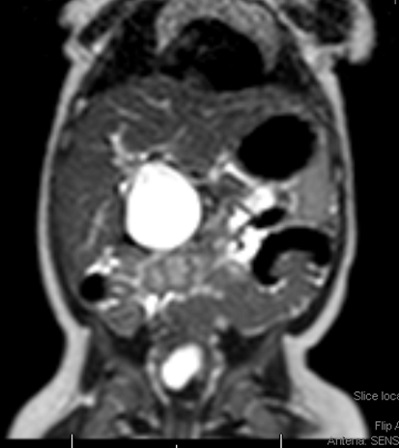
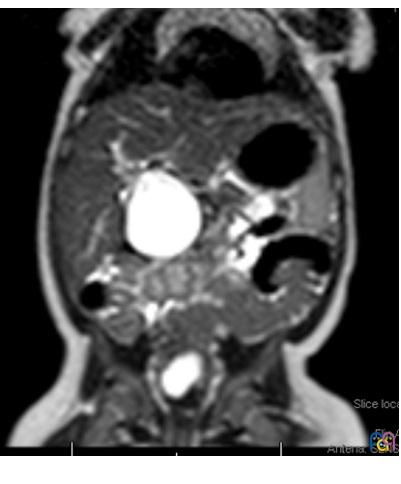
Se realiza ecografía abdominal (Fig. 1), donde se objetiva una marcada dilatación del colédoco, con morfología quístico-fusiforme, que se extiende desde hepático común hasta región pancreática, sugestivo de quiste de colédoco tipo IA según la clasificación de Todani, con barro biliar y leve ectasia de la vía biliar intrahepática. Se completa el estudio con una colangiorresonancia magnética (Fig. 2), donde se visualiza una lesión quística bien definida y redondeada del conducto colédoco en su totalidad, de 4,6 × 3,5 × 3,3 cm, sin extensión a vía biliar intrahepática, confirmando la sospecha diagnóstica inicial.
| Figura 1. Ecografía. Marcada dilatación del colédoco, de morfología quística |
|---|
 |
| Figura 2. Colangiorresonancia magnética. Lesión quística bien definida, amplia, y redondeada del conducto colédoco en su totalidad |
|---|
 |
Se procede a cirugía (cistectomía y hepaticoduodenostomía), con posoperatorio complicado con distensión abdominal y colección en lecho quirúrgico, se realiza laparoscopia en la que no se observa obstrucción intestinal pero sí se sospecha fuga biliar. Se liberan adherencias colohepáticas y se coloca drenaje, con evolución posterior inicialmente favorable.
DISCUSIÓN
El quiste del colédoco es una dilatación congénita de la vía biliar que fue descrita por primera vez por Vater y Ezler en 17231,2. Su etiología es desconocida. El quiste del colédoco se puede asociar con otras anomalías congénitas, como atresia duodenal o colónica, gastrosquisis, páncreas anular y cardiopatías congénitas2.
Aunque pueden diagnosticarse a cualquier edad, el 80% se diagnostica en la primera década de la vida2. La incidencia oscila de 1/13 000 nacidos vivos en Japón hasta 1/100 000 en población occidental1. Es más frecuente en asiáticos y en el sexo femenino, con una predominancia 4:1.
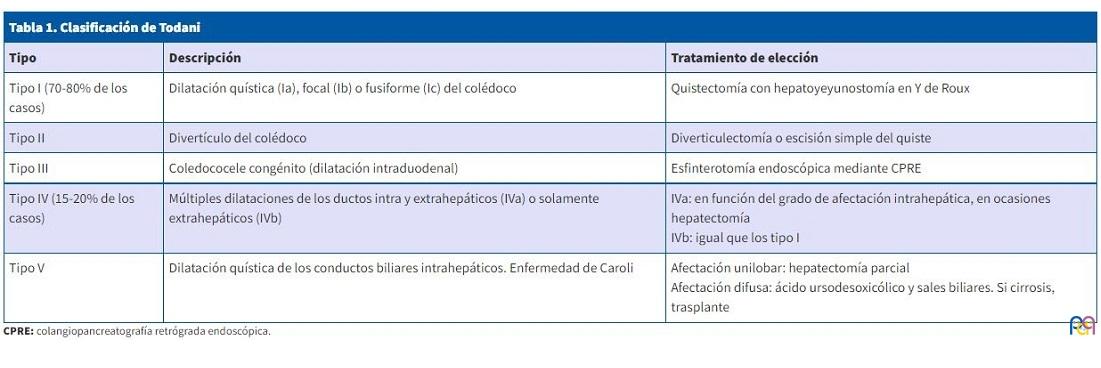
Alonso-Lej et al. publicaron la primera serie de casos de quistes de colédoco en 1959 y formularon la primera clasificación, que fue modificada por Todani et al. en 1977 (Tabla 1)3.
| Tabla 1. Clasificación de Todani | ||
|---|---|---|
| Tipo | Descripción | Tratamiento de elección |
| Tipo I (70-80% de los casos) | Dilatación quística (Ia), focal (Ib) o fusiforme (Ic) del colédoco | Quistectomía con hepatoyeyunostomía en Y de Roux |
| Tipo II | Divertículo del colédoco | Diverticulectomía o escisión simple del quiste |
| Tipo III | Coledococele congénito (dilatación intraduodenal) | Esfinterotomía endoscópica mediante CPRE |
| Tipo IV (15-20% de los casos) | Múltiples dilataciones de los ductos intra y extrahepáticos (IVa) o solamente extrahepáticos (IVb) | IVa: en función del grado de afectación intrahepática, en ocasiones hepatectomía IVb: igual que los tipo I |
| Tipo V | Dilatación quística de los conductos biliares intrahepáticos. Enfermedad de Caroli | Afectación unilobar: hepatectomía parcial Afectación difusa: ácido ursodesoxicólico y sales biliares. Si cirrosis, trasplante |
La presentación clínica es variable. La tríada clásica de ictericia, dolor abdominal y masa abdominal palpable en hipocondrio derecho se da fundamentalmente en la edad pediátrica (el 85% de los niños presenta 2 de las 3 características clínicas, especialmente masa abdominal e ictericia, frente a un 25% de los adultos)4. Por debajo del año de vida se presenta en forma de ictericia, acolia y vómitos. La rotura quística es poco frecuente (1-2%) y ocurre únicamente en neonatos y lactantes1. En niños más mayores o adultos puede no dar síntomas, y hasta en un tercio de los casos su diagnóstico es incidental.
Los hallazgos analíticos son inespecíficos. Puede cursar con hipertransaminasemia, aumento de amilasa, lipasa y leucocitos. La presencia de coagulopatía o alteración de la función renal son marcadores de gravedad.
Para el diagnóstico, es fundamental apoyarse en técnicas de imagen, siendo de elección como prueba inicial la ecografía, que puede detectarlo incluso de forma prenatal1. Pueden emplearse la resonancia magnética o la tomografía computarizada para confirmar la extensión de la afectación ductal o la presencia de afectación extrahepática.
Otras pruebas, como la colangiografía transhepática percutánea o la colangiografía retrógrada endoscópica son más sensibles para definir la anatomía de la vía biliar, pero quedan relegadas a un segundo plano en la edad pediátrica, especialmente por su menor accesibilidad y dificultades técnicas2.
El diagnóstico diferencial incluye la atresia biliar, hepatitis infecciosa, litiasis biliar, pancreatitis, rabdomiosarcoma, colangitis esclerosante primaria, etc.1.
El abordaje específico va a depender del tipo de quiste (Tabla 1), pero de forma general, el objetivo es doble: escisión del quiste y restablecimiento del drenaje bilioentérico de forma directa al duodeno o mediante hepatoyeyunostomía en Y de Roux1,2,4.
La enfermedad sintomática es indicación de cirugía a cualquier edad; sin embargo, en casos de recién nacidos asintomáticos el tiempo quirúrgico es más controvertido, aunque últimamente se tiende a la cirugía más precoz, por debajo del mes de vida en los casos de diagnóstico prenatal, dado que a partir de ahí incrementa la incidencia de fibrosis hepática1,2.
El pronóstico tras la escisión a corto plazo es en general excelente, con un 89% de casos libres de complicación y una supervivencia a los cinco años por encima del 90%. A largo plazo, puede cursar con estenosis anastomótica, colangitis, litiasis y cirrosis. Además, los pacientes con quiste del colédoco tienen un riesgo aumentado de desarrollar colangiocarcinoma y carcinoma de vesícula biliar, incluso tras la cirugía, especialmente los quistes tipo I y IV5. Se desconoce el origen de la progresión cancerígena. En una revisión de 5780 casos, Sastry et al. notificaron un 7.5% de casos de cáncer (colangiocarcinoma [70,4%] y carcinoma de vesícula biliar [23,5%]). La incidencia de malignización antes de los 18 fue de 0,42 frente al 11,4% en adultos6. El pronóstico, una vez desarrollada malignidad, es infausto, con una supervivencia a los 5 años del 5%4. Por este motivo, se recomienda un seguimiento a largo plazo tras la cirugía, con controles analíticos y ecográficos anuales, que además incluyan marcadores cancerígenos como el CA 19-95,7.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
CPRE: colangiopancreatografía retrógrada endoscópica.
BIBLIOGRAFÍA
- Soares KC, Goldstein SD, Ghaseb MA, Kamel I, Hackam DJ, Pawlik TM. Pediatric choledochal cysts: diagnosis and current management. Pediatr Surg Int. 2017;33:637-50.
- Soares KC, Arnaoutakis DJ, Kamel I, Rastegar N, Anders R, Anders R, et al. Choledochal cysts: presentation, clinical differentiation and management. J Am Coll Surg. 2014;219:1167-80.
- Todani T, Wanatabe Y, Narusue M, Tabuchi K, Okajuma K. Congenital bile duct cyst: Classification, operative procedures and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1978;134:263-9.
- Hoilat GJ, John S. Choledochal Cyst. StatPearls Publishing [en línea] [consultado 14/12/2021]. Disponible en www.ncbi.nlm.nih.gov/books/NBK557762/
- Madadi-Sanjani O, Wirth TC, Kuebler JF, Petersen C, Ure BM. Choledochal cyst and malignancy: a plea for lifelong follow-up. Eur J Pediatr Surg. 2019;29:143-9.
- Sastry AV, Abbadessa B, Wayne MG, Steele JG, Cooperman AM. What is the incidence of biliary carcinoma in choledochal cysts, when do they develop, and how should it affect management? World J Surg. 2015;39:487-92.
- Ohashi T, Wakai T, Kubota M, Matsuda Y, Arai Y, Ohyama T, et al. Risk of subsequent biliary malignancy in patients undergoing cyst excision for congenital choledochal cysts. J Gastroenterol Hepatol. 2013;28:243-7.
Comentarios
Este artículo aún no tiene comentarios.