


Bromhidrosis por trimetilaminuria. Dificultades en su diagnóstico y tratamiento
Carmen Sánchez Ortegaa, Regina Rodríguez Martínezb, Rosario Rodríguez Mayatoc, Fernando Sánchez Garcíac
aABS Collblanc. Hospitalet de Llobregat. Barcelona. España.
bUGC Urgencias. Complejo Hospitalario Universitario Torrecárdenas. Almería. España.
cUGC-Pediatría. Hospital La Inmaculada. AGS Norte de Almería. Almería. España.
Correspondencia: C Sánchez. Correo electrónico: carsanort@hotmail.com
Cómo citar este artículo: Sánchez Ortega C, Rodríguez Martínez R, Rodríguez Mayato R, Sánchez García F. Bromhidrosis por trimetilaminuria. Dificultades en su diagnóstico y tratamiento. Rev Pediatr Aten Primaria. 2021;23:e151-e155.
Publicado en Internet: 07-12-2021 - Número de visitas: 13765
Resumen
La trimetilaminuria es una causa de bromhidrosis, que hay que tener en cuenta en niños prepúberes con un desarrollo normal. Su relación con la ingesta, sobre todo de pescado marino, nos hará sospechar su existencia, que deberemos confirmar mediante el análisis de la excreción urinaria de trimetilamina y el estudio genético.
Presentamos cuatro casos de trimetilaminuria donde se analizan y discuten las fórmulas más empleadas para valorar una excreción urinaria aumentada de trimetilamina, su correlación con la gravedad del cuadro y con el rendimiento de las pruebas genéticas.
Por último, se describen los tratamientos empleados y se realiza una propuesta de tratamiento, basada en las recomendaciones dietéticas y en el asesoramiento a los padres para un mejor conocimiento y manejo de la enfermedad.
Palabras clave
● Bromhidrosis ● Niños ● Olor a pescado ● TrimetilaminuriaINTRODUCCIÓN
La bromhidrosis definida como un olor anormal y desagradable, está frecuentemente relacionada con la acción de las bacterias cutáneas sobre las secreciones apocrinas; sin embargo en el niño prepuberal, dichas glándulas apocrinas aún no se han desarrollado, por lo que deberemos plantearnos otras posibles causas; desde un cuerpo extraño en la vía aérea, hasta la ingesta de diferentes tipos de alimentos, pasando por enfermedades metabólicas y enfermedades sistémicas con especial afectación hepática o renal.
Entre las enfermedades metabólicas que cursan sin otro tipo de manifestación clínica más que la bromhidrosis, destaca la trimetilaminuria (TMAU), caracterizada por excreción excesiva de un compuesto maloliente, la trimetilamina (TMA),
La trimetilaminuria (TMAU) en una rara enfermedad metabólica, que afecta a 1 de cada 40 000 personas1 y que se debe a un déficit en la función de la enzima monoxigenasa 3 que contiene flavina (FMO3) encargada de la oxidación hepática de la TMA.
En condiciones normales, el 95% de la TMA procedente del metabolismo intestinal de los alimentos ricos en colina, lecitina y trimetilamina oxidada (TMAO), es oxidada a nivel hepático por la acción de la enzima FMO3, que la transforma en TMAO, compuesto inodoro que se elimina por la orina.
En los pacientes con déficit funcional de la enzima FMO3, al no poder oxidarse a nivel hepático, se va a producir un aumento de la excreción de TMA a través de las secreciones corporales (sudor, orina, etc.)1, provocando un olor corporal que recuerda al pescado en descomposición.
Esta enfermedad fue descrita por primera vez en 1970 por Humbert, y desde entonces se han descrito diferentes formas clínicas; siendo las más frecuentes las formas primarias, las cuales se presentan con un patrón de herencia autosómico recesivo, debido a la mutación del gen FMO3, localizado en el brazo largo del cromosoma 1 (1q24,3). Dicho gen es altamente polimórfico, y se han descrito al menos 40 mutaciones solas o en combinación, que se asocian con una mayor o menor actividad de la enzima FMO31,2.
Las formas secundarias se han relacionado, con patología renal o hepática, fundamentalmente hepatitis, mientras que las formas transitorias, pueden deberse a diferentes causas como: polimorfismos del gen FMO3, inmadurez funcional de la enzima FMO3, excesiva ingesta de precursores de TMA o situaciones especiales relacionadas con el estrés o la menstruación1,3,4.
Ante la sospecha de TMA (basada en la relación directa de los síntomas, con determinados alimentos de la dieta, fundamentalmente pescados marinos), deberemos confirmar dicho diagnóstico mediante el análisis de la excreción urinaria de TMA y TMAO, que demostrará una elevación de la TMA en orina superior al 9%5; y el estudio genético de mutaciones del gen FMO3.
Para aquellos casos sospechosos, con estudio de excreción de TMA en orina normal, se recomienda realizar un segundo estudio, o bien realizar un test de sobrecarga con 300-600 mg de TMA por vía oral y determinación de la excreción de TMA urinaria a las 8-12 horas4,6.
Con análisis genético de las posibles mutaciones del gen FMO32,3, buscaremos la existencia de variantes bialélicas patógenas, relacionadas con la pérdida de función de la enzima FMO33,5,6 habiéndose descrito más de 40 variantes relacionados con un déficit funcional de la enzima.
La mayoría de los tratamientos recomendados están basados en restricciones dietéticas de los precursores de la TMA, lo que incluye alimentos ricos en colina (huevos, hígado, riñones, guisantes, frijoles, maní, soja y brassicas) y ricos en TMAO (pescado de mar, mariscos, cefalópodos y crustáceos).
Dado que la colina es un elemento fundamental en el desarrollo de las membranas celulares, no se aconseja realizar restricciones severas en niños en edad de crecimiento, por el riesgo de ocasionar daños graves a nivel hepático, óseo o del sistema nervioso7,8 sin embargo, sí se pueden realizar sin riesgo, la disminución de alimentos ricos en TMAO8.
También se recomienda el uso de jabones y lociones corporales con pH ácido al objeto de disminuir la volatilidad a nivel cutáneo de la TMA, el uso de resinas de intercambio iónico (carbón activado, y cobre-clorofila; laxantes, y ciclos cortos con antibióticos durante 10 días (neomicina, metronidazol), con el objetivo de modificar la flora intestinal; estos últimos estarían indicados, sobre todo, para situaciones específicas como estrés, menstruación, o determinados actos sociales9.
Por último, también se ha aconsejado el uso de riboflavina, ya que ayudaría a maximizar la actividad de la enzima FMO3. Otros tratamientos en fase de estudio incluyen el uso de probióticos, como modificadores de la flora intestinal y la terapia génica7.
CASOS CLÍNICOS
Presentamos cuatro pacientes, derivados a las consultas externas de un hospital comarcal cuyo motivo de derivación no tenía relación con la bromhidrosis, pero que durante la visita la madre nos consultó por este síntoma, el cual fue percibido, en todos los pacientes, antes de haber cumplido el año de edad. En todos los casos, los padres referían un incremento de la sintomatología relacionado con la ingesta previa de pescado marino, sin otra sintomatología añadida.
El desarrollo pondoestatural y psicomotor de los pacientes fue normal y se descartaron patologías hepáticas, renales y metabólicas incluida diabetes mellitus; así como patologías dermatológicas.
Se determinó la excreción urinaria de TMA y TMAO, la cual se obtuvo sin realizar recomendaciones dietéticas previas.
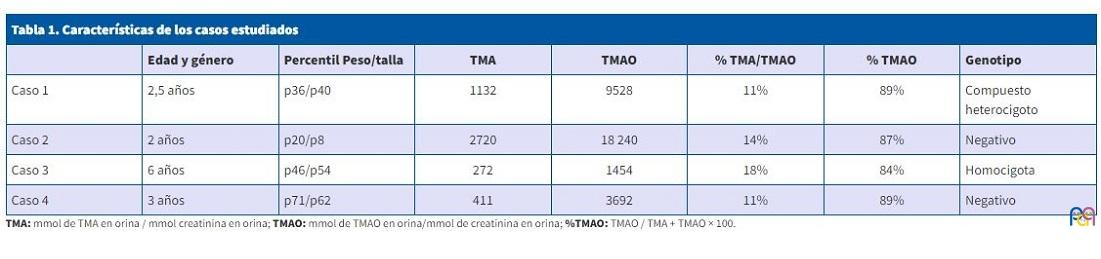
A todos los pacientes se les realizó estudio genético, confirmándose el diagnóstico de sospecha en dos de ellos: casos 1 y 3, con los siguientes resultados: caso 1 compuesto heterocigoto para c.458C>T (p.Prol153 Leu) y c.694G>T, p.Asp232Tyr); caso 3: homocigota para la variante c.472G>A, (p.Glu158Lys) y c.939a>G (p.Glu308Gly). En los otros dos casos no se detectaron variantes patogénicas o de significado clínico incierto para el gen FMO3 (Tabla 1).
| Tabla 1. Características de los casos estudiados | |||||||
|---|---|---|---|---|---|---|---|
| Edad y género | Percentil Peso/talla | TMA | TMAO | % TMA/TMAO | % TMAO | Genotipo | |
| Caso 1 | 2,5 años | p36/p40 | 1132 | 9528 | 11% | 89% | Compuesto heterocigoto |
| Caso 2 | 2 años | p20/p8 | 2720 | 18 240 | 14% | 87% | Negativo |
| Caso 3 | 6 años | p46/p54 | 272 | 1454 | 18% | 84% | Homocigota |
| Caso 4 | 3 años | p71/p62 | 411 | 3692 | 11% | 89% | Negativo |
A todos, se les recomendó tratamiento dietético con restricción parcial del pescado y colina, usando como guía de recomendaciones la historia dietética y la identificación de aquellos alimentos, que provocaban mayor intensidad del mal olor (ver esquema de tratamiento), con buena respuesta al mismo.
En el caso 4, tras dos años de seguimiento, se realizó en dos ocasiones prueba de provocación con dieta libre de pescado y alimentos con mayor contenido en colina con reaparición de la clínica, cada vez que no se seguían las restricciones.
En uno de los casos (caso 3), junto a las medidas dietéticas se recomendó vitamina B2 a dosis de 50 mg/día, con disminución de los episodios de bromhidrosis y la intensidad de estos.
A todos los familiares se les realizó un asesoramiento, para manejo dietético, evitación de desencadenantes, situaciones de riesgo, etc., que fue recibido como de gran utilidad por parte de los padres y ayudó a manejar las situaciones con aumento de la clínica.
DISCUSIÓN
La TMAU es una enfermedad poco frecuente, que debe ser considerada ante la consulta de bromhidrosis en un niño pre púber. La relación con la ingesta de pescado marino y alimentos ricos en colina (carne roja, huevo, frijoles, guisantes,…) nos debe alertar para su diagnóstico. En los casos presentados, llama la atención que, en ninguno, el motivo de derivación fuese la bromhidrosis, cuando este era un problema que preocupaba a sus padres y por el cual ya habían consultado anteriormente en Atención Primaria, lo que podría estar en relación con el desconocimiento de la existencia de dicha patología por su pediatra, dada la escasa prevalencia de la misma.
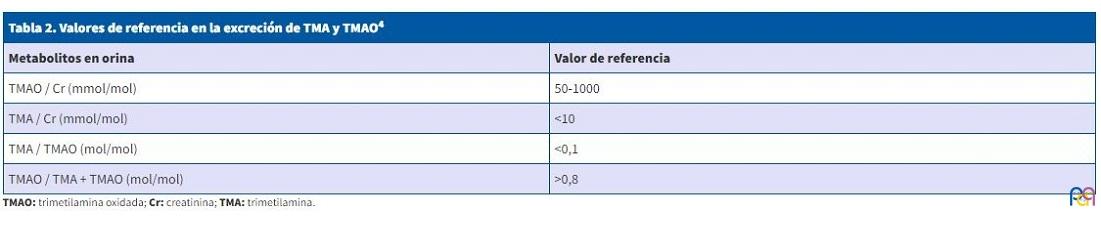
Una de las dificultades encontradas por el clínico para el diagnóstico de TMAU primaria, viene dada por la interpretación de la excreción urinaria de TMA y TMAO, que va a estar influenciada por diversos factores como la dieta, densidad urinaria, flora bacteriana, infecciones o el estrés, y por tanto puede ser muy variable en el tiempo, recomendándose analizar la excreción urinaria de ambos metabolitos en relación a la excreción de creatinina4 (Tabla 2).
| Tabla 2. Valores de referencia en la excreción de TMA y TMAO4 | |
|---|---|
| Metabolitos en orina | Valor de referencia |
| TMAO / Cr (mmol/mol) | 50-1000 |
| TMA / Cr (mmol/mol) | <10 |
| TMA / TMAO (mol/mol) | <0,1 |
| TMAO / TMA + TMAO (mol/mol) | >0,8 |
Para evaluar la excreción urinaria de TMA, usaremos la ratio: [TMAO / (TMA + TMAO)] × 100, idealmente en dos ocasiones distintas, considerándose como afectados aquellos individuos con cifras inferiores a 90-92%5,6.
La gravedad de la afectación esta inversamente relacionada con los resultados de dicha fórmula, de modo que cifras inferiores al 60%, son considerados como formas moderadas-graves de TMAU5.
Existen otras determinaciones que nos pueden ser de utilidad, como la concentración de TMA por mol de creatinina, que en el caso de estar elevada junto con una proporción de TMA/TMAO en orina normal, nos orientará a un exceso de precursores dietéticos de TMA como causantes de dicha elevación; no obstante, teniendo en cuenta la gran variabilidad de la amina liberada en orina, es recomendable calcular todas las proporciones recogidas en la Tabla 24.
Aunque el estudio genético, nos va a dar el diagnóstico definitivo al detectar la mutación en ambos alelos del gen de la FMO3, la existencia de otras posibles mutaciones patológicas, o de dudoso significado, así como de otras formas de TMAU como las formas transitorias o secundarias, hace que el estudio genético pese a ser de utilidad, presente un elevado porcentaje de casos sin detección de alteraciones, sobre todo en casos leves y moderados10; lo que viene a reforzar la importancia del estudio bioquímico de la excreción urinaria de TMA y TMAO, como principal elemento diagnóstico de la TMAU.
Las variantes encontradas en los pacientes de los casos 1 y 3, están descritas en la literatura como variantes que en posición “cis”, se asocian a formas leves-moderadas de TMAU primaria. La no detección de anomalías para el gen FMO3 en dos de los casos, junto a la persistencia de la clínica que se exacerba con la dieta rica en precursores de TMA, podría interpretarse como debida a haplotipos polifórmicos o a otras mutaciones aún no detectadas, o bien a la interrelación ya descrita de formas primarias leves con formas secundarias que puedan causar una disminución ligera de la actividad de la enzima FMO35,6.
Nuestros resultados coinciden con lo descrito en la literatura médica, sobre el bajo rendimiento diagnóstico del estudio genético para los casos de TMA leve, en contraste con su excelente sensibilidad en los casos graves10.
El análisis bioquímico urinario, sigue siendo el método diagnóstico de primera línea para clasificar los diversos tipos de TMAU, siendo probable que el análisis del gen FMO3, solo sea informativo para ciertas presentaciones de TMAU10.
La respuesta al tratamiento fue buena en todos los casos, basándose en la restricción dietética de los desencadenantes de la clínica, fundamentalmente alimentos ricos en TMAO. Para facilitar su manejo, a los padres de nuestros pacientes se les entregó un cuestionario de alimentos que provocan aumento de la TMA (basado en las recomendaciones de Aguilar-Shea et al.8). En dicho cuestionario, los alimentos están clasificados por grupos y los pacientes deben puntuarlos de 0 a 5, según la intensidad de olor que su ingesta les provoca. Se les recomienda no combinar alimentos con puntuaciones altas y dentro de cada grupo, tratar de usar aquellos con menor puntuación.
No se realizó determinación de la excreción de TMA y TMAO tras las restricciones dietéticas o el tratamiento con vitamina B12, lo que hubiese sido recomendable, para poder cuantificar el diferente grado de respuesta a dichas medidas, que presentaron los pacientes5.
Por último, queremos resaltar la gran utilidad, que para los padres supuso el asesoramiento para el manejo de la enfermedad, basado en explicar su causa, los diferentes grados de esta y los posibles desencadenantes de las exacerbaciones. De esta forma, ellos pudieron realizar un manejo de las crisis y comprender el motivo de las mismas. Además, se explicaron los riesgos de las restricciones dietéticas severas, la necesidad de aportar folato en la dieta5, así como los medicamentos cuyo metabolismo puede ser alterado por la deficiencia de FMO3.
CONCLUSIONES
El análisis de la excreción urinaria de la TMA es el método diagnóstico principal sobre todo en formas leves de TMA o formas transitorias y secundarias10.
La fórmula más recomendada para dicho análisis es la relación (TMAO / TMA + TMAO) × 100, considerándose patológicos los valores por debajo del 92%.
El estudio del gen FMO3, pese a estar indicado en todos los casos de TMAU primaria, va a ser de mayor utilidad en los casos moderados o graves (% de TMAO >60%).
Los tratamientos basados en la restricción de determinados alimentos y el conocimiento de los posibles desencadenantes, se consideran el pilar fundamental del tratamiento.
El asesoramiento a los padres ha mejorado el seguimiento y manejo de estos pacientes.
CONFLICTO DE INTERESES
Los autores declaran no presentan conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
FMO3: enzima monoxigenasa 3 que contiene flavina · TMA: trimetilamina · TMAU: trimetilaminuria.
BIBLIOGRAFÍA
- Montoya Álvarez T, Díaz Guardiola P, Olivar Roldán J, Elviro R, Wevers R, Guijarro G. Trimetilaminuria: el síndrome de olor a pescado. Endocrinol Nutr. 2009;56:337-40.
- Romero García A, Bermejo Pastor M, Benito Alonso E, Barros Angueira F, Galán Gómez E. Trimetilaminuria primaria o síndrome del olor a pescado: diagnóstico precoz desde atención primaria. An Pediatr (Barc). 2013;78:272-4.
- Sabir N, Jones EA, Padmakumar B. Trimethylaminuria. BMJ Case Rep. 2016;2015-7.
- Bouchemal N, Ouss l, Brassier A, Barbier V, Gobin S, Hubert l, et al. Diagnosis and phenotypic assessment of trimethylaminuria, and its treatment with riboflavin: 1H NMR spectroscopy and genetic testing. Orphanet J Rare Dis. 2019;14:1-10.
- Phillips IR, Shephard EA. Trimethylaminuria. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Seattle: University of Washington; 2007.
- Mackay RJ, McEntyre CJ, Henderson C, Lever M, George PM. Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin Biochem Rev. 2011;32:33-43.
- Schmidt AC, Leroux JC. Treatments of trimethylaminuria: where we are and where we might be heading. Drug Discov Today. 2020;25:1710-7.
- Aguilar Shea AL, Gallardo Mayo C, Amengual Pliego M, Morais López A. Síndrome del olor a pescado (Trimetilaminuria), la dieta es importante. Rev Esp Nutr Hum Diet. 2016;20:254.
- Chalmers RA, Bain MD, Michelakakis H, Zschocke J, Iles RA. Diagnosis and management of trimethylaminuria (FMO3 deficiency) in children. J Inherit Metab Dis. 2006;29:162-72.
- Doyle S, O’Byrne JJ, Nesbitt M, Murphy DN, Abidin Z, Byrne N, et al. The genetic and biochemical basis of trimethylaminuria in an Irish cohort. JIMD Rep. 2019;47:35-40.
Comentarios
Este artículo aún no tiene comentarios.