

Vol. 21 - Num. 84
Notas clínicas
Nefrocalcinosis, un hallazgo casual, pero un signo guía
Marta Morell Garcíaa, Amparo Ros Floresb, Ana Bayo Pérezc, Nerea Sarrión Sosd, David Aguilera Alonsoa
aServicio de Pediatría. Consorcio Hospital General de Valencia. Valencia. España.
bPediatra. CS Nou Moles. Valencia. España.
cCS Integrado de Alaquás. Hospital General Universitario de Valencia. Valencia. España.
dPediatra. CS Guillem de Castro. Valencia. España.
Correspondencia: M Morell. Correo electrónico: marta.8590@gmail.com
Cómo citar este artículo: Morell García M, Ros Flores A, Bayo Pérez A, Sarrión Sos N, Aguilera Alonso D. Nefrocalcinosis, un hallazgo casual, pero un signo guía. Rev Pediatr Aten Primaria. 2019;21:389-91.
Publicado en Internet: 21-11-2019 - Número de visitas: 13224
Resumen
Se presenta el caso de una niña de diez años diagnosticada de nefrocalcinosis bilateral tras un estudio radiológico solicitado por sospecha de escoliosis. El estudio de sangre y orina inicial nos orienta hacia el diagnóstico de una rara enfermedad, como es el síndrome de hipomagnesemia familiar con hipercalciuria y nefrocalcinosis. El diagnóstico genético dirigido solicitado en ámbito hospitalario confirma el diagnóstico y permite iniciar el tratamiento.
Palabras clave
● Hipercalciuria ● Hipomagnesemia ● NefrocalcinosisCASO CLÍNICO
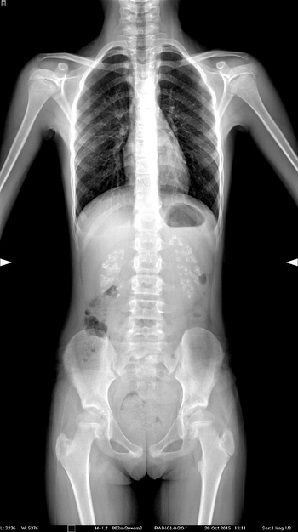
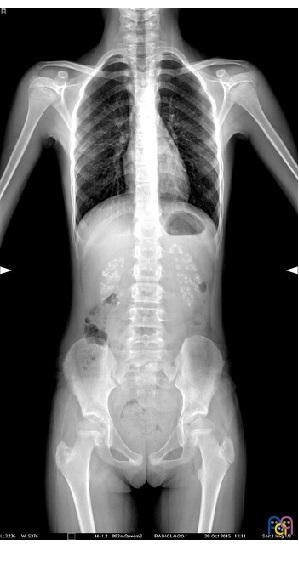
Niña de diez años que acude a la consulta del pediatra de Atención Primaria por dolor lumbar. En la exploración por aparatos se sospecha una escoliosis, el resto de la exploración es normal. Se indica analgesia y se solicita una radiografía de columna. Acude posteriormente para control clínico y resultado de la radiografía, donde se observa como hallazgo incidental nefrocalcinosis bilateral (Fig. 1). En la anamnesis dirigida, refiere polidipsia y poliuria desde la primera infancia, junto a enuresis primaria nocturna resuelta en el último año. Presentaba hasta el momento actual un buen desarrollo psicomotor y ponderoestatural.
| Figura 1. Nefrocalcinosis, hallazgo incidental |
|---|
 |
La paciente no tiene antecedentes personales de interés, pero en ambas ramas familiares se encuentran antecedentes de litiasis renal o enfermedad renal crónica.
En la exploración física, la paciente presenta una presión arterial sistólica en el percentil 95 y diastólica en percentil 75-90, puñopercusión lumbar negativa bilateral y desarrollo puberal en estadio Tanner S2P1A1. Se solicita una analítica de sangre, con resultados de magnesio de 1,4 mg/dl, calcio de 10,1 mg/dl y un filtrado glomerular mediante fórmula modificada de Schwartz de 67 ml/min/1,73 m2. Urea, vitamina D, tirotropina, ácido úrico, fósforo, sodio, potasio, renina, aldosterona, parathormona, gasometría venosa, microalbuminuria y glucosa normales.
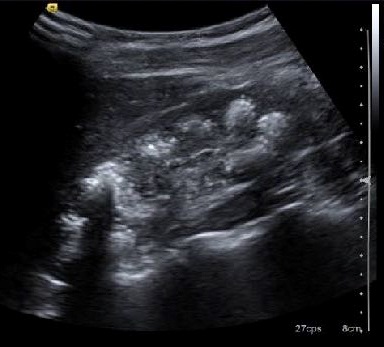
En la consulta externa de Nefrología Pediátrica se completa el estudio con una ecografía renal, que muestra calcificaciones en las pirámides parenquimatosas de ambos riñones sin dilatación (Fig. 2), y orina de 24 horas con la presencia de hipercalciuria (9,9 mg/kg/24 horas) e hipermagnesuria (3,62 mg/kg/24 horas).
| Figura 2. Calcificaciones renales |
|---|
 |

Dada la presencia de hipercalciuria, hipomagnesemia y nefrocalcinosis, se solicita un estudio genético dirigido para el síndrome de hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (FHHNC), siendo portadora homocigota del cambio c.59G>A en el gen CLDN19, el cual nos confirma el diagnóstico.
DISCUSIÓN
El síndrome de hipomagnesemia familiar con hipercalciuria y nefrocalcinosis es una enfermedad rara, de herencia autosómica recesiva provocada por una mutación de los genes CLDN16 y CLDN191.
Dichos genes codifican claudinas, proteínas que intervienen en la reabsorción catiónica a nivel de asa de Henle, produciendo una pérdida renal de calcio y magnesio, con la consecuente hipomagnesemia. El calcio plasmático suele encontrarse en niveles normales por la contrarregulación de la parathormona, la cual se eleva para mantener dichos niveles normales pese a la hipercalciuria2.
El cuadro clínico descrito incluye poliuria, polidipsia, nefrocalcinosis, litiasis renal e infecciones del tracto urinario3. Además, puede afectar a nivel ocular, especialmente si la alteración genética ocurre en el gen CLDN19, con miopía magna, coloboma macular y nistagmo; otros síntomas posibles incluyen, a nivel muscular, tetania debido a la hipomagnesemia y, a nivel del sistema nervioso, convulsiones, neuropatías e inestabilidad en la marcha. También pueden presentar síntomas inespecíficos como vómitos, retraso psicomotor, episodios convulsivos o fallo de medro4. Con todo ello, es importante destacar que se presenta gran variabilidad fenotípica en pacientes con las mismas alteraciones genéticas, y que los pacientes con alteración del CLDN19 presentan mayor progresión a enfermedad renal crónica1.
Por lo dicho, el estudio genético no solo nos proporciona el diagnóstico de certeza, sino que nos aporta cierta información pronóstica y nos permite asesoramiento y consejo genético.
El tratamiento para el FHHNC inicial incluye diuréticos tiazídicos y suplementos de magnesio. Los diuréticos tiazídicos favorecen la disminución de la calciuria y la disminución de la necesidad de suplementos de calcio, pero no induce cambios en el filtrado glomerular, lo que conlleva igualmente a enfermedad renal crónica. Por otro lado, los suplementos de vitamina D y calcio previenen el hiperparatiroidismo secundario5,6.
La evolución propia del síndrome es hacia enfermedad renal crónica, apareciendo en un tercio de los casos en las primeras dos décadas de vida, precisando finalmente trasplante renal que resulta curativo5.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS
FHHNC: síndrome de hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (familial hypomagnesaemia with hypercalciuria and nephrocalcinosis).
BIBLIOGRAFÍA
- Claverie-Martín F, Vargas-Poussou R, Müller D, García-Nieto V. Clinical utility gene card for familial hypomagnesemia with hypercalciuria and nephrocalcinosis with/without severe ocular involvement. Eur J Hum Genet. 2015;23:889.
- Rönnefarth G, Misselwitz J. Nephrocalcinosis in children: a retrospective survey. Pediatr Nephrol. 2000;14:1016-21.
- Martín Aguado M, Canals Baeza A, Sanguino López L, Gavilán Martín C, Flores Serrano J. Hipomagnesemia familiar con hipercalciuria y nefrocalcinosis. An Pediatr (Barc). 2001;54:174-7.
- Claverie-Martín F. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: clinical and molecular characteristics. Clin Kidney J. 2015;8:656-64.
- Sikora P, Zaniew M, Haisch L. Retrospective cohort study of familial hypomagnesaemia with hypercalciuria and nephrocalcinosis due to CLDN16 mutations. Nephrol Dial Transplant. 2015;30:636-44.
- Shroff R, Aitkenhead H, Costa N, Trivelli A, Litwin M, Picca S, et al. Normal 25-hydroxyvitamin D levels are associated with less proteinuria and attenuate renal failure progression in children with CKD. J Am Soc Nephrol. 2016;27:314-22.
Comentarios
Este artículo aún no tiene comentarios.