


Manejo terapéutico de la atresia de vías biliares
David Crehuet Gramatykaa, Gemma Navarro Rubioa
aCorporación Sanitaria y Universitaria Parc Taulí. Facultad de Medicina. Unidad Docente Parc Taulí. Universidad Autònoma de Barcelona. Barcelona. España.
Cómo citar este artículo: Crehuet Gramatyka D, Navarro Rubio G. Manejo terapéutico de la atresia de vías biliares. Rev Pediatr Aten Primaria. 2016;18:e141-e147.
Publicado en Internet: 12-09-2016 - Número de visitas: 16697
Resumen
La atresia de vías biliares es una enfermedad de origen desconocido que causa ictericia neonatal. Es la primera causa de trasplante hepático en niños (40%). Esto es debido a que su técnica correctora, la portoenterostomía de Kasai (KPE), únicamente consigue el drenaje biliar en aproximadamente un 60% de los casos. A pesar del correcto drenaje, solo un 20% llega a edad adulta con su hígado nativo, y una gran parte de ellos con complicaciones hepáticas y calidad de vida subóptima. Estos datos hacen replantearse continuamente el manejo terapéutico y si el trasplante podría ser el tratamiento de elección. El objetivo de esta revisión es actualizar la información disponible con el fin de determinar la mejor estrategia en su manejo.
Partiendo de la base bibliográfica ya seleccionada, se realizó una búsqueda con los términos “biliary atresia” y “treatment”. Debido a que el trasplante hepático a edades tempranas implica peores resultados, actualmente se prefiere la KPE como primera elección y el trasplante como tratamiento de rescate.
Palabras clave
● Atresia biliar ● TratamientoINTRODUCCIÓN
La atresia de vías biliares (AVB) es una enfermedad fibroobliterativa y progresiva de etiología desconocida que afecta las vías biliares intra- y extrahepáticas y causa ictericia neonatal por obstrucción grave del flujo biliar. Aparece en uno de cada 10 000-15 000 nacidos vivos1,2, predominando en el sexo femenino (4:1) y en la raza asiática1,2. Constituye la causa más común de trasplante hepático infantil, siendo esta etiología la responsable del 40% de las indicaciones de trasplante1.
La etiología es desconocida, aunque se sabe que no es una enfermedad hereditaria. El 20% de casos se asocian a otras malformaciones1,2. El cuadro clínico típico es el de un recién nacido a término sano, con fenotipo y peso normal, que desarrolla ictericia progresiva, coluria y acolia en las dos primeras semanas de vida por falta de drenaje de la bilis. Suele mostrar hepatomegalia y signos de hipertensión portal, mostrando una evolución hacia la cirrosis y la insuficiencia hepática. Estos niños, dejados a su evolución, no sobreviven más de dos años1.
Una vez que se sospecha el diagnóstico de AVB, este se refuerza con una ecografía en la que en ocasiones se ve ausencia o disminución del tamaño de la vesícula biliar, y con una gammagrafía hepatobiliar con tecnecio-99, que muestra ausencia de flujo biliar hacia el duodeno. Para hacer el diagnóstico definitivo es necesario realizar una colangiografía.
Según el patrón colangiográfico, se clasifica en tres tipos:
- Atresia tipo I: obliteración del colédoco, con libre tránsito por los conductos proximales.
- Atresia tipo II: obstrucción del conducto hepático, pero a nivel del hilio hepático hay dilatación quística de los conductos. Subtipos: IIa) libre tránsito por los conductos cístico y colédoco, y IIb) obliteración de ambas estructuras.
- Atresia tipo III (corresponde al 90% y también la de peor pronóstico): no se identifican conductos hepáticos o hilio dilatado. Últimamente se postula que este tipo no es una enfermedad congénita, sino que es una obliteración fibrótica del recién nacido de causa desconocida.
El tratamiento de esta entidad es muy discutido, ya que los resultados a largo plazo no son muy buenos por ineficiencia de la técnica quirúrgica correctora, la portoenterostomía de Kasai (KPE). El objetivo de este trabajo es revisar los conocimientos respecto el manejo terapéutico y los resultados en los pacientes afectados para determinar la mejor estrategia en el manejo de la patología.
BÚSQUEDA BIBLIOGRÁFICA
Partiendo de la base bibliográfica recogida en el texto de A. Coran1, se realizó una búsqueda en PubMed con los términos MeSH “biliary atresia” y “treatment”. Se incluyeron los artículos publicados posteriormente al 2005 y escritos en inglés. Se obtuvieron 2446 artículos; se excluyó la mayoría por no adaptarse a la temática propuesta. Finalmente, se analizaron 19 estudios.
TRATAMIENTO DE LA AVB
El manejo terapéutico de la AVB es muy controvertido, ya que cerca del 80-85% de los que padecen esta enfermedad acabarán siendo trasplantados1,2,4,5.
Hoy en día se considera la portoenterostomia de Kasai (KPE) como la primera línea de tratamiento. No obstante, este es un tema controvertido, ya que la mayoría de artículos defienden las ventajas de esta intervención como primera línea1,2,4,5,7,8 frente algún artículo de reciente aparición que apuesta por el trasplante como primera opción3.
Portoenterostomía de Kasai
La KPE consiste en una operación donde se diseca el remanente fibrótico hasta el espacio porta, se corta a ras de este y se anastomosa un asa intestinal yeyunal para que sirva de conducto de drenaje a la bilis, restableciendo el flujo alimentario mediante una anastomosis yeyuno-yeyunal a unos 30-40 centímetros del borde yeyunal anastomosado al hígado1,2.
Preoperatorio
En el preoperatorio de esta cirugía está indicado administrar antibiótico preoperatorio y suplemento vitamínico liposoluble. No suele ser necesaria la limpieza intestinal prequirúrgica y se recomienda un ayuno de 4 horas, para asegurar un buen vaciamiento gástrico sin exponer el lactante al riesgo de hipoglucemia1,2.
Resultados de portoenterostomía y trasplante hepático
La KPE se considera la primera línea tratamiento para esta entidad, siendo en muchos casos un puente al trasplante, excepto cuando el paciente presente insuficiencia hepática. En este caso la primera opción terapéutica sería el trasplante hepático, igual que si existen malformaciones asociadas (las más frecuentes esplénicas), pues se ha visto que en estos casos la portoenterostomía tiene mayores índices de fracaso1,2,4,6,7.
También se han descrito atresias asociadas a infección por citomegalovirus (CMV), que suelen debutar en edad más tardía y tienen peor pronóstico, por lo cual se considera también el trasplante como primera elección6.
El objetivo de esta técnica es crear un sistema de drenaje de la bilis y conseguir una disminución de la ictericia. No obstante, solo aproximadamente un 60% de los pacientes consiguen tasas libres de bilirrubina en el postoperatorio (Tabla 1). En el 40% restante no se consigue un correcto drenaje y requieren trasplante hepático.
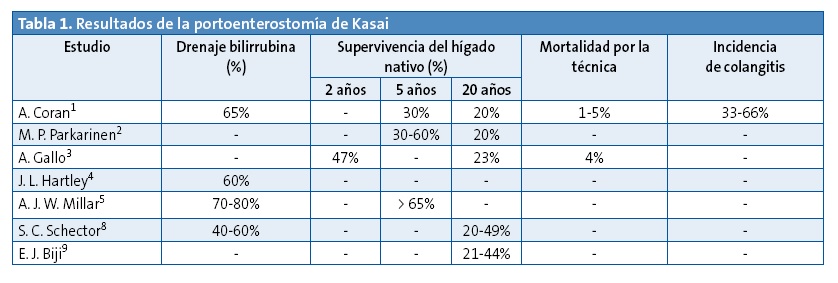
El motivo por el cual no se consigue un correcto drenaje suele deberse a la fibrosis de la anastomosis, que puede estar presente en el momento de la anastomosis y corresponderá a ese 40% de pacientes que no consiguen drenar la bilis. De los pacientes que consiguen drenar y disminuir su bilirrubina, unos presentarán reobstrucción precoz y otros reobstrucción tardía. Estos pacientes presentarán empeoramiento de la función hepática progresiva hasta ser necesaria la realización de un trasplante.
Podemos observar que la KPE tiene un 1-4% de mortalidad y una supervivencia con hígado nativo a los 20 años del 20%, lo que crea controversia sobre si el trasplante podría ser considerado como técnica de primera elección.
Las razones de los defensores del trasplante son:
- Mejoría de los resultados en trasplantes hepáticos debido a un avance científico en todas las áreas del trasplante3 (procedimientos, conservación del órgano, immunosupresores, etc.) (Tabla 2).
- Según el estudio de A. Gallo3, la supervivencia del KPE y el trasplante es similar, mientras que las complicaciones infecciosas serán mayores en el primer caso. Si comparamos resultados de calidad de vida encontramos que el 86% de los pacientes con KPE tienen trabajo frente al 62% en trasplantados, y educación superior el 71 frente al 63%, aunque estos resultados se pueden ver influidos por el hecho de que los pacientes trasplantados suelen ser los más graves.
- El trasplante es un tratamiento definitivo.
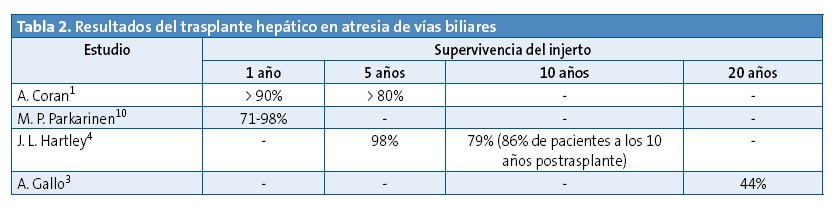
A pesar de las mejorías en el trasplante, la gran mayoría de estudios apuestan por la KPE como opción de primera línea1,2,4,5,7,8. Los motivos principales son: los resultados del trasplante mejoran a medida que avanza la edad del niño1,11,12 (trasplantar a menores de 10 kg tiene peor pronóstico) y los efectos nocivos de someter a niños de edades tan tempranas a los efectos del trasplante (tratamientos inmunosupresores, etc.).
Un estudio reciente ha demostrado que en los pacientes a los cuales se realiza KPE y presentan fallo tardío, esta tiene efecto protector para la supervivencia del trasplante hepático (presentan un 55% menos de posibilidades de ser retransplantados y un 84% menos de fallecer) respecto a los trasplantados directamente. Los pacientes que presentan fallo precoz tienen los mismos resultados en el trasplante que en los que no se realizó KPE, pero tienen más incidencia de complicaciones biliares y perforaciones intestinales12 (Tabla 3).
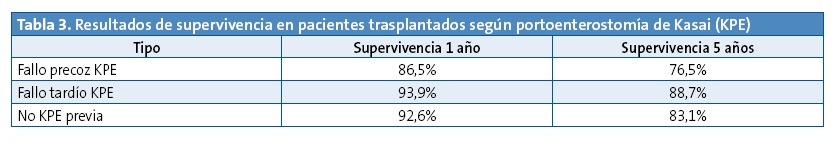
Por tanto, se sigue considerando el KPE como tratamiento de primera línea, pero es necesaria una constante evaluación de los resultados debido al avance del trasplante12.
No obstante, debido al desconocimiento de la causa de la enfermedad que cursa con esta fibrosis, y a los insatisfactorios resultados de la cirugía, la gran mayoría de la comunidad científica considera necesario la investigación de la etiología de la enfermedad. Esta dará paso a nuevas terapias que permitan detener la fibrosis y obtener mejores resultados a largo plazo3,4,7,12.
Factores pronósticos
Los factores que se asocian a mejores resultados en la KPE y por tanto a mayor supervivencia con hígado nativo son:
- Edad del paciente: todos los estudios avalan que es necesario la realización de la KPE lo más precozmente posible, puesto que ello mejora mucho los resultados y es un factor crucial1,2,5,7,8,10,11. Se considera que se debe intervenir antes de los 90 días. Después de esta fecha los resultados empeoran sustancialmente y se recomienda proceder directamente al trasplante, aunque, dependiendo de si el estado basal del niño es bueno, se puede optar por la portoenterostomía10. Otros estudios muestran resultados son mejores si se practica la cirugía antes de los 30 días de vida7.
- Macroanatomía de la vía biliar: las atresias tipo I y II (10% de los pacientes) se asocian a mejor pronóstico, debido a que tienen un remanente de vía biliar permeable. Estos tipos son los que más se beneficiaran del KPE, por lo cual es importante una correcta identificación de estos pacientes ya que probablemente no requerirán trasplante1,5,7,10. Por el contrario, la KPE tipo III (el 90% de los pacientes) es la que ofrece peores resultados y mayores tasas de fracaso del KPE.
- El grosor de los ductos biliares después de la disección del espacio porta: (es necesario obtener una biopsia para el estudio anatomopatológico) es un buen predictor de fracaso de la técnica, ya que, si los ductos son superiores a 150 micras, la tasa de éxito es del 86%, mientras que si son inferiores desciende hasta el 12%1,5,10.
- Experiencia del cirujano: mayor experiencia se asocia a mejores resultados1,5. Se ha observado que los centros especializados, con mayor número de casos, tienen mejores resultados1,10, aunque algunos estudios que lo ponen en duda por falta de estudios reglados5.
- Estado previo del hígado: un peor estado previo produce mayores tasas de fracasos1,5.
- Colangitis posoperatorias: dan peores resultados5.
Variantes de la KPE
Con el desarrollo de la laparoscopia se ha propuesto utilizar este método para la KPE. No obstante, en todos los estudios se ha visto que no ofrece ninguna ventaja sobre la laparotomía1,5,6,10,13. No hay diferencias en el tiempo operatorio, la estancia hospitalaria, la pérdida de sangre intraoperatoria, la desaparición de la ictericia precoz o la tasa de colangitis. Se encontró una mayor supervivencia a 20 años en pacientes tratados con la laparotomía frente a laparoscopia13. La única ventaja que se ha observado es la disminución de fibrosis perihepática de cara al trasplante10.
Se han propuesto técnicas alternativas a la KPE clásica. A pesar de ello, todas se están desaconsejadas ya que no aportan beneficios y, en algunos casos, pueden presentar mayor riesgo de complicaciones1,5. La única que ha demostrado utilidad es la gallblader Kasai portoenterostomy para la atresia tipo II5; pero esta, de por sí, tiene buen pronóstico.
Existe una modificación de la KPE que consiste en realizar una sutura más superficial en los conductos hepáticos, que ha demostrado una mayor supervivencia tanto en su variante laparoscópica como por laparotomía respecto a la KPE clásica por laparotomia16. Sería conveniente realizar más estudios para validar su eficacia.
Postoperatorio
Es muy importante el postoperatorio. Como medidas profilácticas tenemos:
- Fenobarbital: disminuye la producción de bilirrubina5,12. No obstante, hay un artículo que critica su uso por falta de eficacia demostrada6.
- Suplementación con vitaminas liposolubles1,5,12.
- Ácido ursodesoxicólico: mejora la fluidez biliar1,5,6,12.
- Fórmulas de lactancia especiales a partir del tercer día1,6,12.
- Corticoides: es un tema muy controvertido, pues desde siempre se ha postulado que por su efecto antiinflamatorio producían un mejor drenaje biliar, pero últimamente se ha descubierto que no aumentan la supervivencia1,2,5,6,10,12,14,15 ni contribuyen al descenso de la bilirrubina14,15, aunque hay otros estudios que dicen que sí2,10. No obstante, todos ellos concluyen que son necesarios más estudios para determinar su eficacia.
- 48 horas de sonda nasogástrica1.
- Profilaxis con trimetoprim-sulfametoxazol1.
Complicaciones de la KPE
Las complicaciones más frecuentes de la KPE son:
- Colangitis: es la complicación más frecuente y se debe al ascenso de las bacterias por vía ascendente1. Un 60% la presentará en el transcurso de cinco años19.
- Déficit metabólico y nutricional: algunos autores sugieren como indicación de trasplante el déficit nutricional o de crecimiento ya que proporciona un mayor crecimiento1,4.
- Fibrosis e hipertensión portal (HTP): las complicaciones de HTP son la principal causa de trasplante post-KPE1,12.
- Ictericia en el 55% de los pacientes17 y alteración de la función hepática.
La mayoría de las complicaciones son debidas al fallo de la anastomosis enteroportal, por eso se ha sugerido como tratamiento la reoperación de Kasai, también llamada REDO surgery. La evidencia científica no avala esta práctica, pues se asocia a peores resultados en el trasplante, aunque puede ser considerada en caso de falta de injerto para el transplante18. No obstante, existe un estudio que demuestra que, en pacientes que restablecieron el flujo biliar pos-KPE pero que se estancó durante el primer mes posoperatorio, son candidatos a beneficiarse de la REDO surgery20.
Resultados a largo plazo de la KPE
Aproximadamente el 20% de los pacientes operados llegan a adultos con su hígado nativo. No obstante, solo el 53% de estos pacientes refiere su calidad de vida como normal, y un 1,8% como ideal19. Esto es debido a que entre el 70-98% de pacientes desarrollan fibrosis a pesar de la KPE y alteración de la función hepática entre el 73-85%6,19.
El 10% de los niños sufren retraso de crecimiento y un 40% prurito crónico17. Según un estudio de seguimiento de pacientes con hígados nativos, solo tres de 14 mujeres que buscaban embarazo lo consiguieron (es muy importante seguir a estas pacientes durante el embarazo, pues tienden a presentar mayores complicaciones). En resumen, algunos pacientes sobreviven con hígado nativo pero a expensas de enfermedad biliar residual, colangitis e hipertensión portal.
CONCLUSIÓN
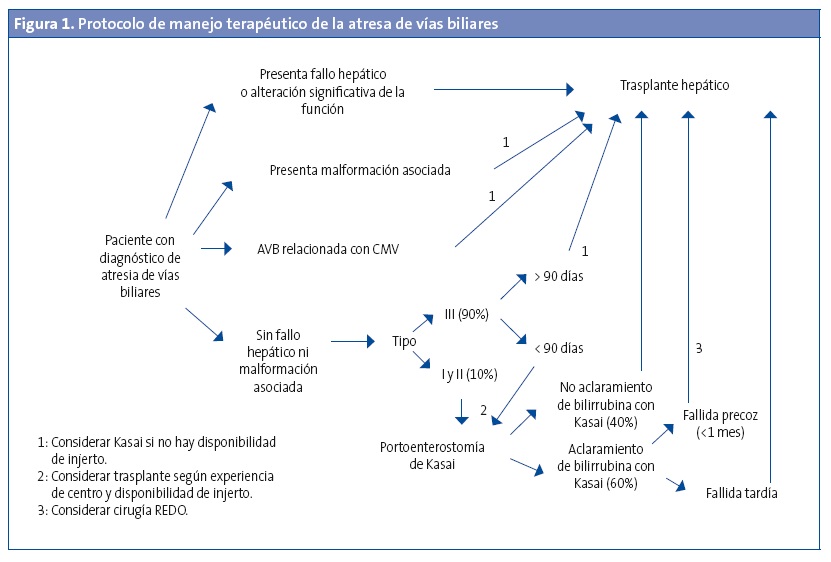
La atresia de vías biliares es una enfermedad de difícil tratamiento, puesto que la técnica de primera elección, la portoenterostomía de Kasai (KPE), tiene muchos fracasos. Para aquellos en los que se logra restituir el flujo biliar, tiene muchas complicaciones y una calidad de vida subóptima. Con la información hoy disponible, el mejor tratamiento consiste en individualizar cada caso (Fig. 1), pero en la mayoría de casos se considera como primera línea tratamiento la KPE por el hecho de que algunos pacientes puedan preservar su hígado nativo y evitar los efectos adversos del trasplante hepático y porque esta técnica pueda ser un puente al trasplante a una edad más tardía. Es importante un correcto manejo interdisciplinar (cirujanos pediátricos, trasplantadores, digestólogos, pediatras, etc.) y seguir revisando constantemente los resultados con nuevos estudios. Los esfuerzos en investigación deben ir principalmente dirigidos a entender el proceso fisiopatológico de la enfermedad y así encontrar nuevas dianas (probablemente moleculares) de tratamiento.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS: AVB: atresia de vías biliares · CMV: citomegalovirus · HTP: hipertensión portal · KPE: portoenterostomía de Kasai.
BIBLIOGRAFÍA
- Coran AG, Adzick NS, Krummel TM, Laberge JM, Shamberger R, Caldamome A. Pediatric surgery. 7.ª edición. Filadelfia: Elsevier; 2012.
- Pakarinen MP, Rintala RJ. Surgery of biliary atresia. Scand J Surg. 2011;100:49-53.
- Gallo A, Esquivel CO. Current options for management of biliary atresia. Pediatr Transplant. 2013;17:95-8.
- Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009:374;1704-13.
- Millar AJ. Solving difficult hepatobiliary problems in children. S Afr Med J. 2012;102:872-5.
- Davenport M, Grieve A. Maximizing Kasai portoenterostomy in the treatment of biliary atresia: medical and surgical options. S Afr Med J. 2012;102:865-7.
- Jimenez-Rivera C, Jolin-Dahel KS, Fortinsky KJ, Gozdyra P, Benchimol EI. International incidence and outcomes of biliary atresia. J Pediatr Gastroenterol Nutr. 2013;56:344-54.
- Schecter SC, Courtier J, Cho SJ, Saadai P, Hirose S, Mackenzie TC, et al. Hepatic portocholecystostomy for biliary atresia: a 25-year follow-up and review. J Pediatr Surg. 2013;48:262-6.
- Bijl EJ, Bharwani KD, Houwen RH, de Man RA. The long-term outcome of the Kasai operation in patients with biliary atresia: a systematic review. Neth J Med. 2013;71:170-3.
- Davenport M. Biliary atresia: clinical aspects. Semin Pediatr Surg. 21 2012. EE. UU.: Elsevier Inc; 2012. p. 175-84.
- Neto SJ, Feier FH, Bierrenbach AL, Toscano CM, Fonseca EA, Pugliese R, et al. Impact of Kasai portoenterostomy on liver transplantation outcomes: a retrospective cohort study of 347 children with biliary atresia. Liver Transpl. 2015;21:922-7.
- Hadzic N. Medical management of the 'failing' Kasai portoenterostomy. S Afr Med J. 2012;102:868-71.
- Lishuang M, Zhen C, Guoliang Q, Zhen Z, Chen W, Long L, et al. Laparoscopic portoenterostomy versus open portoenterostomy for the treatment of biliary atresia: a systematic review and meta-analysis of comparative studies. Pediatr Surg Int. 2015;31:261-9.
- Zhang D, Yang HY, Jia J, Zhao G, Yue M, Wang JX. Postoperative steroids after Kasai portoenterostomy for biliary atresia: a meta-analysis. Int J Surg. 2014;12:1203-9.
- Sarkhy A, Schreiber RA, Milner RA, Barker CC. Does adjuvant steroid therapy post-Kasai portoenterostomy improve outcome of biliary atresia? Systematic review and meta-analysis. Can J Gastroenterol. 2011;25:440-4.
- Wada M, Nakamura H, Koga H, Miyano G, Lane GJ, Okazaki T, et al. Experience of treating biliary atresia with three types of portoenterostomy at a single institution: extended, modified Kasai, and laparoscopic modified Kasai. Pediatr Surg Int. 2014;30:863-70.
- Kumagi T, Drenth JP, Guttman O, Ng V, Lilly L, Therapondos G, et al. Biliary atresia and survival into adulthood without transplantation: a collaborative multicentre clinic review. Liver Int. 2012;32:510-8.
- Urahashi T, Ihara Y, Sanada Y, Wakiya T, Yamada N, Okada N, et al. Effect of repeat Kasai hepatic portoenterostomy on pediatric live-donor liver graft for biliary atresia. Exp Clin Transplant. 2013;11:259-63.
- Ng VL, Haber BH, Magee JC, Miethke A, Murray KF, Michail S, et al. Medical status of 219 children with biliary atresia surviving long-term with their native livers: results from a North American multicenter consortium. J Pediatr. 2015;166:211.
- Nio M, Sasaki H, Tanaka H, Okamura A. Redo surgery for biliary atresia. Pediatr Surg Int. 2013;29:989-93.
Comentarios
Este artículo aún no tiene comentarios.