

Vol. 17 - Num. 68
Revisiones
Déficit de glucosa-6-fosfato deshidrogenasa: revisión a propósito de un caso
Pablo Bello Gutiérreza, L Mohamed Dafab
aServicio de Pediatría. Hospital Universitario Rey Juan Carlos. Móstoles. Madrid. España.
bPediatra. Servicio Madrileño de Salud. España.
Correspondencia: P Bello. Correo electrónico: pablo.bello@hospitalreyjuancarlos.es
Cómo citar este artículo: Bello Gutiérrez P, Mohamed Dafa L. Déficit de glucosa-6-fosfato deshidrogenasa: revisión a propósito de un caso. Rev Pediatr Aten Primaria. 2015;17:361-8.
Publicado en Internet: 23-11-2015 - Número de visitas: 52647
Resumen
El déficit de glucosa-6-fosfato deshidrogenasa (DG6PDH) es el defecto enzimático más frecuente de los eritrocitos. Se trata de una alteración vinculada a la protección del glóbulo rojo frente al estrés oxidativo. La mayoría de los pacientes están asintomáticos. Clínicamente, se asocia con cuadros de hemólisis, desencadenada por algunos fármacos, infecciones o alimentos. En el caso de asociarse a la la ingesta de habas, se denomina favismo. El tratamiento está enfocado hacia la anemización producida, precisando en algunos casos trasfusión de hematíes. El principal cuidado de estos pacientes es el de evitar los desencadenantes conocidos de la hemólisis. El D6GPDH no se ha relacionado con una disminución de la calidad o compromiso de la vida de estos pacientes. Se presenta en este artículo una revisión del tema a propósito de un caso que debuta desde Atención Primaria.
Palabras clave
● Anemia hemolítica ● Déficit de glucosa-6-fosfato deshidrogenasa ● Favismo ● Habas ● MalariaINTRODUCCIÓN
El favismo es una de las formas clínicas de presentación del déficit de glucosa-6-fosfato deshidrogenasa (DG6PDH), que es el defecto enzimático de los glóbulos rojos más frecuente en los seres humanos, y que afecta a cerca de 400 millones de personas en el mundo1.
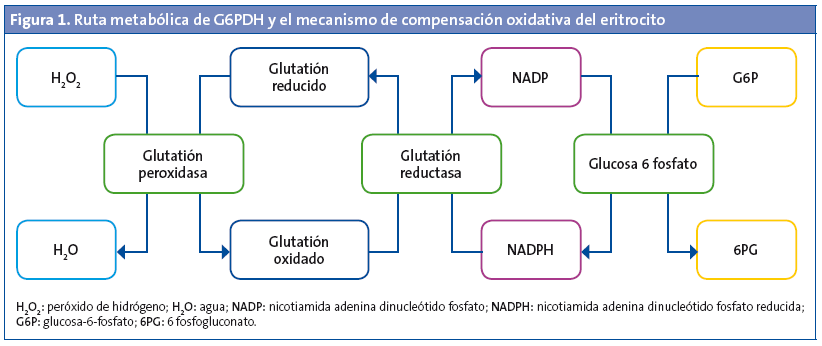
La glucosa-6-fosfato deshidrogenasa (G6PDH) es un enzima eritrocitaria cuya función consiste en mantener la homeostasis de los eritrocitos frente a los insultos oxidativos, a través de la producción de nicotinamida adenina dinucleótido fosfato reducida (NADPH). Esta enzima forma parte de la ruta metabólica de las pentosas monofosfato y cataliza el paso oxidativo de la glucosa-6-fosfato hacia 6-fosfogluconato y reduce la nicotinamida adenina dinucleótido fosfato (NADP) a NADPH (Fig. 1). Esta vía provee de NADPH al eritrocito, y es un cofactor básico en el metabolismo del glutatión, que participa activamente en la protección frente a estímulos oxidativos. El eritrocito tiene de forma habitual una gran cantidad de glutatión reducido que actúa como amortiguador de noxas endógenas o exógenas (infecciones, medicamentos y algunos alimentos). De esta manera no se produce el acúmulo oxidativo ni la degeneración proteica eritrocitaria, debido al paso del glutatión oxidado a reducido, para lo que se emplea NADPH, que a su vez se acopla a la actividad de la G6PDH. El eritrocito depende activamente de la producción de NADPH por esta vía para el balance del estrés oxidativo, al no disponer de mitocondrias para su obtención1,2.
CASO CLÍNICO
Son valorados en el centro de salud dos gemelos monocigotos de tres años por un cuadro de distermia sin termometrar de 24 horas de evolución, dolor abdominal, vómitos, orinas oscuras, color amarillo de la piel y debilidad generalizada. En la exploración física llamaba la atención el marcado decaimiento, la ictericia, así como una hepatomegalia de 1 cm. Son remitidos a Urgencias hospitalarias para descartar hepatitis aguda.
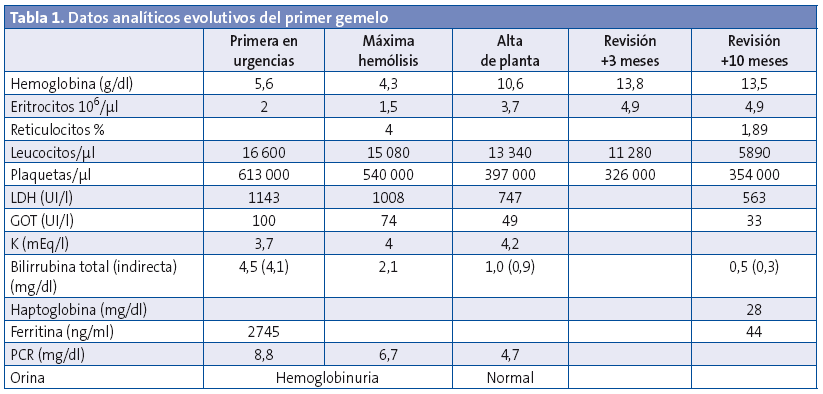
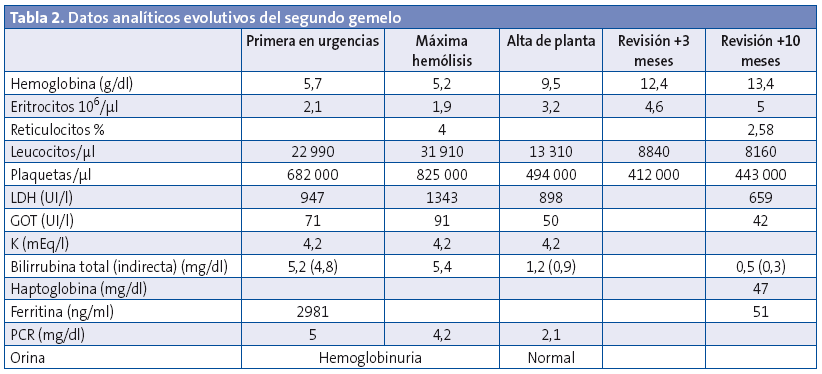
En el hospital se rehistoria a la familia, que no refieren antecedentes de interés, ni ingesta de alimentos nuevos en los días previos, uso de medicaciones o un estado febril. En la exploración física se constata el decaimiento y la ictericia, así como una hepatomegalia leve. Tensión arterial de 76/41 mmHg y 79/53 mmHg, con frecuencias cardiacas de 143-150 latidos por minuto (lpm). Normosaturación. Afebriles en ese momento. En el sistemático de orina se observa hemoglobinuria. Se extrae hemograma y bioquímica de sangre, en la que se objetiva anemia con elevación de las cifras de bilirrubina, LDH y GOT (Tablas 1 y 2). Coombs negativo. Frotis de sangre periférica sin hallazgos específicos salvo lisis. Diuresis espontánea y taquicardia sostenida de hasta 180 lpm. En los siguientes controles analíticos progresa la anemización, por lo que se decide trasfundir un concentrado de hematíes. Se inició terapia intravenosa con cefotaxima por la afectación del estado general con distermia (suspendida a las 48 horas, tras comprobar la esterilidad de los cultivos y por la evolución), así como fluidoterapia con bicarbonato para conseguir alcalinización de la orina como factor de protección renal. Son ingresados en la planta. Con las medidas iniciadas se remontan las cifras de hemoglobina, hasta estabilización de la crisis lítica en las 48 horas posteriores al ingreso. Se extrae estudio completo de anemia hemolítica no inmune.
La madre admite posteriormente un cuadro suyo similar al actual cinco años antes, tras ingesta de habas. Rehistoriada la familia, admiten ingesta de las mismas tres días antes del inicio del cuadro.
Tras estabilizar la crisis lítica, se inicia tratamiento con ácido fólico y seguimiento en consultas para completar el estudio etiológico.
PATOGENIA. ASPECTOS GENÉTICOS
El déficit de esta enzima, en presencia de determinados oxidantes, aumenta la vulnerabilidad de los glóbulos rojos a la destrucción, ya que no son capaces de revertir la acción oxidativa. Si bien el mecanismo exacto de cómo el estrés oxidativo produce hemólisis no se conoce, el efecto es la desnaturalización de la hemoglobina y la disminución de la vida media de los hematíes2.
La G6PDH está presente en todas las células, variando su concentración según los tejidos. En los glóbulos rojos sanos, esta enzima funciona al 1-2% de su capacidad. Esto provee una idea del potencial reductor que se pierde cuando existe un DG6PDH.
El DG6PDH presenta un patrón de herencia ligado al cromosoma X. El gen ligado a esta alteración está en el brazo largo de este cromosoma (Xq28). Los varones son hemicigotos para esta alteración, pudiendo ser normales en la actividad enzimática o deficitarios. En el caso de las mujeres, debido al efecto Lyon, pueden ser heterocigotas (comportándose como mosaicos) y en ellas se han descrito casos clínicos similares en comportamiento a los del varón hemicigoto. Las mujeres homocigotas no son raras en poblaciones con alta incidencia de deficiencias alélicas de G6PDH3.
El DG6PD es uno de los trastornos enzimáticos con mayor heterogeneidad genética. Se han descrito más de 140 mutaciones, con más de 400 variantes bioquímicas del enzima. De acuerdo a la actividad enzimática y las manifestaciones clínicas la Organización Mundial de la Salud (OMS) las ha clasificado en cinco clases que se resumen a continuación4:
- Clase I: su prevalencia es poco frecuente y normalmente el nivel de deficiencia es grave que se manifiestan como anemia hemolítica no esferocítica, o anemia crónica en presencia de función eritrocítica normal.
- Clase II: más prevalente en el Mediterráneo y Asia. Aquí el nivel de deficiencia también es grave y la actividad enzimática es menor del 10% de lo normal.
- Clase III: presente en el 10% de los varones negros de Estados Unidos. Incluye variantes con nivel de deficiencia moderado y una actividad enzimática del 10 al 60% de lo normal.
- Clase IV: es una variante rara donde la deficiencia enzimática suele ser leve o ninguna, y el nivel de actividad enzimática del 60 al 150% del normal.
- Clase V: no hay deficiencia enzimática, también es rara en cuanto a prevalencia y la actividad enzimática es mayor al 150% de lo normal.
CLÍNICA
Dada la gran heterogeneidad genética, la forma de presentación clínica también es bastante variable. La mayoría de los pacientes con este déficit suelen estar asintomáticos.
Las formas clínicas sintomáticas son:
- Anemia hemolítica aguda (por fármacos o infecciones). Existe una larga lista de fármacos y agentes infecciosos que se han relacionado con cuadros de hemólisis aguda. En el caso de fármacos, la hemólisis no es clínicamente detectable hasta las 24-72 horas de su administración. El rasgo característico es el de orina oscura por hemoglobinuria. La anemia se agudiza hasta los 7-8 días de la administración, momento en el que la hemoglobina inicia la recuperación (ver referencias a fármacos relacionados más adelante). En el caso de infecciones, se ha descrito para los virus A y B de la hepatitis, citomegalovirus, neumonías, fiebre tifoidea y agentes como E. coli, Salmonella y Streptococcus grupo B2. El mecanismo exacto de hemólisis por esta causa es desconocido, aunque una explicación podrían ser las reacciones derivadas de la actividad fagocitaria en el seno de la infección.
- Favismo. Aunque la evidencia clínica no lo ha demostrado del todo, la mayoría de los autores coinciden en que la patogenia está determinada por la toxicidad que producen elementos del haba, como la vicina y la convicina, al ser hidrolizadas en el tubo digestivo y convertirse en alguno de los activos divicina e isouramilo, que son capaces de producir hemólisis de los glóbulos rojos6.
- Anemia hemolítica congénita no esferocítica. Es una forma de hemólisis crónica, agrupada en el tipo I de la OMS. Son casos esporádicos. Se trata de una hemólisis típicamente extravascular, que se debe sospechar por una historia compatible (típicamente ictericia neonatal, anemia crónica regenerativa que se exacerba con estímulos oxidativos, colelitiasis, esplenomegalia), así como datos analíticos de destrucción corpuscular.
- Hiperbulirrubinemia neonatal. Es más típica y grave en pretérminos que en términos. Se suele presentar entre los días 1 y 4 de vida, como la fisiológica, y con las mismas complicaciones y tratamiento. El mecanismo de la hemólisis no se conoce totalmente. Se debe pensar en este defecto en el caso de ictericias en las primeras 24 horas, con valores altos, o en casos de historia familiar previa al nacimiento.
De las dos variantes más frecuentes, la G6PD A- (clase III de la OMS) afecta fundamentalmente a africanos y sus descendientes. Su sintomatología en general es poco grave debido a que solo un 20-30% de los eritrocitos deficientes sufren hemólisis. Se suele presentar con poca frecuencia como favismo. La forma mediterránea (G6PD mediterránea), que se considera la clase II de la OMS, afecta fundamentalmente a poblaciones de origen griego, italiano, español, árabe, como el caso de nuestros pacientes, y judíos. Esta variante produce además hiperbilirrubinemia neonatal más grave y el favismo con mucha mayor frecuencia.
Los pacientes con DG6PDH tienen mayor vulnerabilidad para la sepsis y las complicaciones relacionadas con la misma.
Favismo
Se conoce desde el siglo XX, y aunque inicialmente se identificaba como propio de países mediterráneos, se ha ido extendiendo a medida que este producto se ha ido consumiendo en otros países. Se piensa que es la forma más frecuente asociada con la variante mediterránea del DG6PDH, pero deben existir múltiples factores individuales, ya que no todos los pacientes desarrollan síntomas ante su ingesta o en relación a la cantidad tomada.
Está asociado a la ingesta de habas frescas crudas, frescas o secas cocinadas, por inhalación durante el paseo en el campo de habas, a través de la leche materna o animal que haya ingerido habas o también por el contacto con la henna7.
Como rasgos característicos, los síntomas aparecen tras las 5-24 horas de la ingesta con los siguientes elementos diferenciales: hemoglobinuria más grave que en otras formas clínicas, bilirrubina con valores menos elevados que en otras formas del déficit, anemia aguda y grave (que en ocasiones precipita fallo renal agudo por isquemia o depósitos de hemoglobina). En ocasiones hay esplenomegalia.
MANEJO DE LOS PACIENTES
Si bien la estrategia más eficaz en el DG6PDH es la prevención de la hemólisis, a través de la evitación del desencadenante, esta aproximación precisa del conocimiento del defecto por el paciente y su familia. Esto implica que solo cuando se ha tenido alguna crisis previa se es consciente de esta situación.
En los casos de debut, o de transgresiones con los desencadenantes, la sintomatología dependerá de la intensidad de la anemia y la velocidad de su instauración. El manejo, por tanto, se hará desde el enfoque de las anemias hemolíticas.
La historia inicial debe recoger aspectos básicos:
- Antecedentes personales:
- Edad, sexo y etnia: el DG6PDH se asocia a varones por estar ligado al X, en poblaciones de raza negra y zona endémicas de malaria.
- Evolución de los síntomas para valorar la cronicidad o no del cuadro.
- Episodios similares previos, en casos ya conocidos.
- Síntomas infecciosos acompañantes o en días previos como posibles desencadenantes.
- Fármacos que toma o que ha tomado recientemente, o ingesta de habas.
- Aspecto de la orina.
- Antecedentes familiares: historia familiar de anemia, litiasis biliar, esplenectomía (anemias hemolíticas hereditarias). Casos de episodios similares en la familia.
Se debe realizar una exploración física dirigida a la identificación de la repercusión de la crisis hemolítica (que es la forma más frecuente) y sus manifestaciones: coloración de piel y mucosas (palidez, ictericia); frecuencia cardiaca y presión arterial (taquicardia, hipotensión); auscultación cardiopulmonar (soplos); datos clínicos de fallo cardiaco (edemas, hepatomegalia, hipotensión arterial, dificultad respiratoria); hepato/esplenomegalia.
Las formas crónicas son menos sintomáticas.
Exploraciones complementarias
Ante un cuadro de crisis hemolítica aguda se debe remitir a un centro hospitalario para estudios, valoración y tratamiento:
- Hemograma + reticulocitos + pruebas cruzadas + Coombs directo.
- Frotis en sangre periférica. Cuando es normal orienta hacia un defecto de membrana específico.
- Bioquímica de sangre. Se observará:
- Elevación en la LDH, bilirrubina total y fraccionada y GOT.
- Perfil renal: Valora datos de insuficiencia renal por daño agudo de metabolitos de la hemoglobina.
- Haptoglobina: baja (<30 mg/dl).
- Orina: Hemoglobinuria, urobilinógeno.
- Si hay clínica infecciosa concomitante:
- Serologías infecciosas si existen datos compatibles: virus de Epstein Barr, citomegalovirus, virus herpes simple, virus varicela-zóster, virus de la inmunodeficiencia humana, hepatitis, Mycoplasma.
- Cultivos: frotis faríngeo para bacterias/virus; coprocultivo; cultivos de sangre y orina; gota gruesa si procede de área endémica de malaria.
Los estudios complementarios ante una crisis hemolítica para el diagnóstico final específico estarán en función de la orientación del cuadro clínico y la sospecha. En el caso del DG6PDH, se realiza un estudio enzimático específico para determinar la actividad enzimática. La caracterización completa de la enzima solo se realiza en pocas ocasiones (definición de una nueva variante, estudios poblacionales, casos raros y necesidad de diagnóstico prenatal)2.
TRATAMIENTO DE LAS CRISIS HEMOLÍTICAS AGUDAS EN EL DG6PDH
- Medidas generales:
- Ingreso hospitalario en todos los casos para control estrecho. Se valorará la necesidad de cuidados intensivos si no hay control de la hemólisis, con necesidad de trasfusiones frecuentes, o si hay inestabilidad hemodinámica mantenida tras trasfusión de concentrado de hematíes.
- Retirada del agente externo desencadenante en caso de fármacos y alimentos sospechosos.
- Tratamiento antibiótico específico si hay síntomas infecciosos compatibles con infección bacteriana.
- Hidratación y alcalinización de la orina si datos hay de hemólisis intravascular:
- Suero glucosado al 5%: necesidades basales + 50%. No añadir potasio.
- Bicarbonato sódico 1 M: 30-40 mEq/l (para mantener pH urinario entre 7-8) como medida de protección renal.
- Ácido fólico: un comprimido de 5 mg/día mientras dure la crisis hemolítica.
- Transfusión de concentrado de hematíes (CH)(15-20 ml/kg): está indicado siempre que exista alteración hemodinámica y si las cifras de hemoglobina (Hb) <7 g/dl. Si la hemoglobina está entre 7-9 g/dl:
- Si persisten datos de hemólisis intravascular activa (hemoglobinuria): transfusión de CH.
- Si no hay datos de hemólisis intravascular activa: observación con controles clínicos frecuentes y analíticos (8-24 horas).
- Controles clínico-analíticos en fase aguda: control de la anemia y respuesta medular (hemograma con bioquímica con LDH cada 6-8-12-24 horas y reticulocitos cada 24-48 horas), control de la hemoglobinuria con tira de orina en cada micción, así como control clínico exhaustivo con constantes por turno de enfermería.
LA VIDA CON DG6PDH. CONSIDERACIONES DEL MANEJO AMBULATORIO
Una vez que el paciente ha sido diagnosticado de DG6PDH y superada la etapa de la crisis hemolítica, se plantea el manejo posterior. En el caso del favismo, dado que estos pacientes no presentan dependencia transfusional, no es necesario establecer un seguimiento concreto. En los casos de DG6PDH con curso crónico debería seguirse un control periódico, por el riesgo de crisis hemolítica.
Según la Sociedad Española de Hematología y Hemoterapia (SEHH), los pacientes deben saber que la estrategia más eficaz en el manejo de esta entidad es la prevención de la hemólisis. Deberían estar informados sobre cómo evitar los factores asociados al estrés oxidativo. Deben evitar una serie de medicamentos9 y en caso de necesitar de analgesia, se recomienda paracetamol.
En cuanto a la alimentación, en el caso del favismo pueden seguir un régimen normal, salvo la ingesta de habas o productos que las contengan, como purés o potajes, que quedan prohibidos. También se recomienda evitar el contacto con la planta del haba.
Estos pacientes deberían acudir a su médico ante cuadros infecciosos, aunque sean banales, con el objetivo de valorar el inicio de una crisis hemolítica. Si padecen enfermedades crónicas como la diabetes, deberán ser estrechamente vigilados.
Por su parte, la American Family Physician plantea que además de que los pacientes con DG6PD deberían evitar la exposición a los medicamentos que pueden desencadenar una crisis hemolítica y la ingesta de habas, también hay que hacer cribado neonatal de DG6PD en presencia de ictericia neonatal cuando hay antecedentes familiares conocidos de este déficit y según la procedencia geográfica y étnica1.
El DG6PDH no parece afectar a la esperanza de vida, la calidad de la misma o la actividad diaria de los afectados. Afortunadamente, muchos de los pacientes con esta alteración están asintomáticos a lo largo de su vida, completamente ajenos a su condición2.
Los pacientes y sus familias pueden encontrar recursos en las asociaciones de pacientes con DG6PDH (no en español), como:
- Association Française des personnes atteintes du déficit génétique en G6PD ou Favisme (VIGIFAVISME) (Francia): www.vigifavisme.com.
- Associazione Italiana Favismo - Deficit di G6PD (Italia): www.g6pd.org/it.
- Volwassenen, Kinderen en Stofwisselingsziekten (VKS) (Países Bajos): www.stofwisselingsziekten.nl.
- The Purine Metabolic Patients' Association (PUMPA) (Reino Unido): www.pumpa.org.uk.
DISCUSIÓN
A pesar de ser el defecto corpuscular enzimático más frecuente en el ser humano, el DG6PDH es una entidad poco frecuente en la práctica clínica en Pediatría. Datos obtenidos de la población adulta estiman la prevalencia de este defecto entre un 0,1-0,5% de la población (hasta el 0,6% en Andalucía, Extremadura, País Vasco y Baleares)7.
La forma de debut habitual es como un cuadro hemolítico agudo, en el que se llega al origen corpuscular enzimático tras la exclusión del componente inmune y la normalidad de los eritrocitos en la extensión de sangre periférica. El diagnóstico se concluye tras estudios de funcionalidad de la enzima afectada.
Lo infrecuente en Pediatría de esta condición hace que el cuadro sea difícil de identificar. Si bien inicialmente los pacientes llegaron al centro hospitalario para descartar una hepatitis aguda, tras la realización de los primeros estudios se enfocó el caso como una anemia hemolítica, con Coombs directo negativo. La estabilización inicial requirió, tal y como se recoge en la revisión, la trasfusión de un concentrado de hematíes, debido a la extensa destrucción corpuscular. La crisis lítica cedió en las primeras 48 horas, en cuyo contexto se pudo iniciar el estudio etiológico, habiendo cuenta del antecedente familiar tan claro en la madre. Por tanto, es clave en muchas ocasiones la historia suministrada por la familia para el diagnóstico. La sospecha de favismo era alta, dados los antecedentes familiares y la historia actual, en la que no es habitual un cuadro de anemia hemolítica en dos gemelos de forma concomitante.
Los pacientes siguieron en revisión de forma ambulatoria, con controles analíticos que demuestran una recuperación sin recaída de la hemólisis. Diez meses después del ingreso y la trasfusión se hace un estudio enzimático que pone de manifiesto una disminución de la actividad de la G6PDH. Permanecen estables, sin nuevas intercurrencias y con restricción de habas en la dieta, y sin nuevas crisis hemolíticas ante procesos intercurrentes.
La evolución analítica de ambos pacientes se puede seguir en las tablas (Tablas 1 y 2).
CONCLUSIONES
El DG6PDH es un trastorno enzimático que afecta a los glóbulos rojos, ocasionando en los casos sintomáticos crisis hemolíticas o hemólisis crónica. Se calcula que al menos 400 millones de personas en todo el mundo presentan este déficit, el 7% de la población mundial según la OMS, con alta prevalencia en África, Asia, Medio Oriente y la cuenca del Mediterráneo4.
La mayoría de los casos con esta deficiencia están asintomáticos a lo largo de su vida. La clínica suele desencadenarse en presencia de determinados factores asociados al estrés oxidativo, como las infecciones, la ingesta de habas o algunos medicamentos y productos químicos. Por su variabilidad genética, la sintomatología puede ser muy leve, como ocurre en la clase G6PD A- (predominante en África), hasta anemia hemolítica grave, como en el caso del favismo o forma mediterránea clase II de la clasificación de la OMS.
Salvo en los casos que cursan con hemólisis grave que requieren de transfusión y algunas formas crónicas, los pacientes no requieren control médico, y pueden llevar una vida normal, aunque sí conviene que estén informados sobre su enfermedad y sobre los aspectos relacionados con el desencadenamiento de las crisis hemolíticas. Asimismo, estos pacientes deberían acudir a su médico ante la presencia de infecciones banales para valorar el inicio de una crisis hemolítica.
Por último, cabe destacar que, aunque en España esta patología no es frecuente, como se comentaba previamente, podría estar influenciada por los flujos migratorios, sobre todo los procedentes de la zona del Magreb.
CONFLICTO DE INTERESES
Los autores declaran no presentar conflictos de intereses en relación con la preparación y publicación de este artículo.
ABREVIATURAS: CH: concentrado de hematíes • DG6PDH: déficit de glucosa-6-fosfato-deshidrogenasa • G6PDH: glucosa-6-fosfato-deshidrogenasa • NADP: nicotinamida-adenina-dinucleótido-fosfato • NADPH: nicotinamida-adenina-dinucleótido-fosfato reducido • OMS: Organización Mundial de la Salud • SEHH: Sociedad Española de Hematología y Hemoterapia.
BIBLIOGRAFÍA
- Frank JE. Diagnosis and management of G6PD deficiency. Am Fam Physician. 2005;72:1277-82.
- Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64-74.
- Hsia YE, Miyakawa F, Baltazar J, Ching NS, Yuen J, Westwood B, et al. Frequency of glucose-6-phosphate dehydrogenase (G6PD) mutations in Chinese, Filipinos, and Laotians from Hawaii. Hum Genet. 1993;92:470-6.
- WHO Working Group. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ. 1989;67:601-11.
- Luzzatto L. Genetics of red cells and susceptibulity to malaria. Blood. 1979;54:961-76.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol. 1990;27:1-40.
- Verdugo P, Calvanese M, Rodríguez D, Cárcamo C. Deficiencia de glucosa 6 fosfato deshidrogenasa en niños. Caso clínico. Rev Chil Pediatr. 2014;85:74-9.
- Del Lujan Acosta I, Milani AC, Pérez SM, Lanza O, Detarsio G. Deficiencia de glucosa-6-fosfato deshidrogenasa eritrocitaria en Rosario. Acta Bioquím Clín Latinoam. 2012;46:359-63.
- El déficit de glucosa-6-fosfato deshidrogenasa (G6PD). El favismo. Sociedad Española de Hematología y Hemoterapia [en línea] [consultado el 27/10/2015]. Disponible en http://eritropatologia.com/portal/wp-content/uploads/2012/05/AEHH-DG6PDH.pdf
Comentarios
Este artículo aún no tiene comentarios.